《物理化学实验》课程
课程编号:
实验指导书
主撰人 王书媚
审核人 刘 鑫
单位:澳门十大赌博正规官网
二O一二年 五 月
目 录
实验一
| 恒温水浴的组装及其性能测试
| 6
|
实验二
| 燃烧热的测定……
| 9
|
实验三
| 碳酸钙分解反应热的测定
| 14
|
实验四
| 溶解热的测定
| 16
|
实验五
| 纯液体饱和蒸气压的测定
| 21
|
实验六
| 双液系气—液平衡相图
| 25
|
实验七
| 二组分金属相图的绘制
| 29
|
实验八
| 差热分析
| 32
|
实验九
| 化学反应平衡常数和分配系数的测定
| 36
|
实验十
| 分解反应平衡常数的测定
| 39
|
实验十一
| 原电池电动势的测定
| 42
|
实验十二
| 希托夫法测定离子迁移数
| 46
|
实验十三
| 镍在硫酸溶液中的钝化行为
| 49
|
实验十四
| 电导法测定乙酸的电离常数
| 54
|
实验十五
| 铁氰化钾在玻碳(金、铂)电极上的氧化还原
| 57
|
实验十六
| 电导法测定乙酸乙酯皂化反应速率常数
| 61
|
实验十七
| 旋光法测定蔗糖水解反应速率常数
| 65
|
实验十八
| 丙酮碘化反应速率方程的测定
| 68
|
实验十九
| H2O2催化分解反应速率常数的测定
| 72
|
实验二十
| 最大泡压法测定溶液的表面张力
| 75
|
实验二十一
| 固体在溶液中的吸附
| 80
|
实验二十二
| 表面活性剂CMC值的测定
| 82
|
实验二十三
| B-Z振荡反应
| 85
|
实验二十四
| 溶胶聚沉值的测定
| 89
|
实验二十五
| 溶液吸附法测定比表面积
| 92
|
实验二十六
| 电渗
| 96
|
实验二十七
| Fe(OH)3胶体电动电位的测定—界面电泳法
| 98
|
实验二十八
| 粘度法测定水溶性高聚物的分子量
| 101
|
实验二十九
| 配合物磁化率的测定
| 106
|
前 言
1.实验总体目标
物理化学实验是继大学物理实验、无机化学实验、分析化学实验、有机化学实验之后独立开设的又一门重要必修实验课程,是化学专业实验教学体系中的重要组成部分,是物理化学理论课程的延伸。它利用物理学的方法和手段,综合了化学领域中各分支所需的基本研究方法和实验工具,研究物质的物理性质及其与化学变化和相变化之间的关系。
物理化学实验有两大特点:第一,物理化学实验主要是大量使用仪器或若干仪器组成一个实验系统对某个或若干个物理化学性质进行测定,进而研究化学问题。因而实验技术(主要是使用仪器的能力)在物理化学实验中是十分重要的。随着实验技术的不断更新和发展,实验仪器性能朝着快速、准确、便捷的方向发展,因而物理化学实验的手段和方法也必然不断更新、不断发展。第二,由于物质的物理化学性质和化学反应性能往往是间接测量得到的,直接测量的结果还需利用数学的方法加以整理和综合运算,才能得到所需的结果,因此物理化学实验具有能培养综合实验能力和科学研究能力的特点。
本课程教学额总体目标是:巩固并加深对物理化学基本概念和理论的理解,掌握物理化学实验的基本方法、实验技术及常用仪器的构造、原理和使用方法,学会正确观察记录实验现象和数据、选择实验条件、处理实验数据、分析实验结果、撰写实验报告,培养节约、卫生、环保的良好习惯和认真、严谨、求实的科学精神,为今后开展专业提高实验、毕业论文及独立科学研究打下坚实的基础。
2.适用专业年级
化学本科专业三年级
3.实验课时分配
实验项目
| 实验要求
| 实验类型
| 每组人数
| 实验
学时
|
实验一恒温水浴的组装及其性能测试
| 必做
| 验证
| 3
| 5
|
实验二 燃烧热的测定
| 必做
| 验证
| 3
| 5
|
实验三碳酸钙分解反应热的测定
| 选做
| 验证
| 3
| 5
|
实验四溶解热的测定
| 选做
| 验证
| 3
| 5
|
实验五纯液体饱和蒸气压的测定
| 必做
| 验证
| 3
| 5
|
实验六双液系气—液平衡相图
| 必做
| 验证
| 3
| 5
|
实验七二组分金属相图的绘制
| 必做
| 验证
| 3
| 5
|
实验八差热分析
| 必做
| 验证
| 3
| 5
|
实验九化学反应平衡常数和分配系数的测定
| 选做
| 验证
| 3
| 5
|
实验十分解反应平衡常数的测定
| 必做
| 验证
| 3
| 5
|
实验十一 原电池电动势的测定……………
| 选做
| 验证
| 3
| 5
|
实验十二 希托夫法测定离子迁移数
| 选做
| 验证
| 3
| 5
|
实验十三 镍在硫酸溶液中的钝化行为
| 选做
| 验证
| 3
| 5
|
实验十四 电导法测定乙酸的电离常数
| 选做
| 验证
| 3
| 5
|
实验十五 铁氰化钾在玻碳(金、铂)电极上的氧化还原
| 选做
| 验证
| 3
| 5
|
实验十六 电导法测定乙酸乙酯皂化反应速率常数
| 必做
| 验证
| 3
| 5
|
实验十七 旋光法测定蔗糖水解反应速率常数
| 必做
| 验证
| 3
| 5
|
实验十八 丙酮碘化反应速率方程的测定
| 选做
| 验证
| 3
| 5
|
实验十九 H2O2催化分解反应速率常数的测定
| 选做
| 验证
| 3
| 5
|
实验二十 最大泡压法测定溶液的表面张力
| 必做
| 验证
| 3
| 5
|
实验二十一 固体在溶液中的吸附
| 选做
| 验证
| 3
| 5
|
实验二十二 表面活性剂CMC值的测定
| 必做
| 验证
| 3
| 5
|
实验二十三 B-Z振荡反应
| 必做
| 验证
| 3
| 5
|
实验二十四 溶胶聚沉值的测定
| 选做
| 验证
| 3
| 5
|
实验二十五 溶液吸附法测定比表面积
| 选做
| 验证
| 3
| 5
|
实验二十六 电渗
| 必做
| 验证
| 3
| 5
|
实验二十七
Fe(OH)3胶体电动电位的测定—界面电泳法
| 必做
| 验证
| 3
| 5
|
实验二十八粘度法测定水溶性高聚物的分子量
| 选做
| 验证
| 3
| 5
|
实验二十九 配合物磁化率的测定
| 选做
| 验证
| 3
| 5
|
4.实验环境
物理化学学科现有实验教学用房面积150平方米,包括基础部分的物理化学实验室、综合化学实验室、药品准备室、教师实验准备室、大型仪器室、天平室、教师科研用房等;可以开设20多个基础物理化学实验。目前的仪器设备均有与实验教材要求相匹配,便于教学。大多数实验项目配6-8套实验仪器。
5.实验总体要求
物理化学实验总体要求以智能开发、能力培养为核心。具体地说就是物理化学实验教学在重视知识、技能学习的同时,更要重视研究能力的培养,把教学过程很好的结合起来。在前期要求学生做好规定的实验,这些实验包括热力学、动力学、电化学、表面胶体和大分子化学,以及物质结构等分支的典型实验,熟悉其相应的实验方法、实验技术和实验仪器。这是整个物理化学实验的核心,通过实验操作这一中心环节,为培养能力打好基础。在后期,根据情况适当安排一些不同类型的“研究式实验”:重做或改进一些老实验,选作或设计一些新实验。一般说来,这些实验由老师给定题目,自己提出方案,并独立完成配制和标定溶液,组装仪器,以及测量和数据处理等。学生应写出试验报告,并进行交流和总结。
总体要求还包括实验讲座。实验讲座是达到实验目的的重要补充环节。讲座分两部分:一是物理化学实验的一些基本知识、技能,如实验数据的表达与作用,实验结果的误差分析,实验数据的处理方法等,要放在实验操作训练开始之前进行,因为这些基本技能在每一个实验报告中都要用到,对于这些基本技能的训练和严格要求,要贯穿于整个物理化学实验教学的始终。二是物理化学实验的一些基本实验方法和技术,如温度的测量和控制,压力的测量和校正,真空技术,光学测量技术,电化学测量技术等,这些要在学生实验操作训练的基础上,分阶段进行,以开阔学生解决实际问题的能力。
6.本课程的重点、难点及教学方法建议
本课程采用启发式实验教学方法,要求学生在实验前预习有关实验内容,通过自学和与教师之间的主动交流,掌握实验的原理、步骤、数据处理方法等。教师实验前预讲重点改为实验的设计思想及应用,以拓宽学生的知识面。在强调基本操作训练的同时,给学生以独立操作和独立思考的空间。
实验一 恒温水浴的组装及其性能测试
一、实验目的
1.了解恒温水浴的构造及其工作原理,学会装配恒温水浴装置;
2.测绘恒温水浴的灵敏度曲线;
3.掌握数显贝克曼温度计及数显恒温水浴装置的使用方法。
二、实验主要设备及使用要求
恒温水浴装置一套,数显贝克曼温度计
超级恒温槽使用步骤:工作室水箱应注入适量的洁净自来水,加热管至少应低于水面1cm,把
搅拌磁子放入水箱中央,将控温旋钮调到最低,接通电源,把“设定/测量”开关置于“设定”端观察显示屏,调节控温旋钮,调至所需的设定温度,然后回到“测量”。当设定值高于所测定温度时,加热开始工作。显示屏显示为探头所测的实际温度。当加热到所需的温度时,加热会自动停止,低于设定的温度时,新的一轮加热又会开始,为了保证水温均匀性,应打开搅拌开关,慢慢调节调速旋钮。
三、实验原理、方法和手段
恒温水浴是一种常用的控温装置,由浴槽(盛蒸馏水)、温度传感器、电子继电器、温度设定装置、温度计、加热器和搅拌器组成。当温度传感器感受到浴槽的水温(T)后,就会将电信号传给温度设定装置,如果实验中我们通过温度设定装置所设定的温度(也就是我们希望恒温水浴保持恒定的温度)为T',那么,在经过对比之后,电子继电器就会对加热器发出加热或停止加热的指令来控制水温。
当恒温水浴的温度T因散热等原因使水浴温度低于设定值T'时,电子继电器就指示加热器工作,使恒温水浴的温度上升。由于加热过程中温度传感器、温度设定装和电子继电器一直处于相互通讯的工作状态,随着恒温水浴的温度T的不断升高,当T达到设定值T'时,电子继电器就立即指示加热器停止加热。这样周而复始地进行加热、停止加热、再加热、再停止加热的操作,理论上可以使恒温水浴的温度始终保持在T'的状态。
但是,在恒温水浴装置的实际运行过程中,由于热量传递需要时间,以及加热器的供热速度与恒温水浴的散热速度的不断变化的缘故,恒温水浴的温度不可能始终保持在T'的状态而不发生变化,而会出现如下的描述那样的周期性变化。
当T由于加热而刚好等于T'时,即使加热器接到电子继电器的指令而停止工作了,但加热器还在继续散发余热,使恒温水浴的散热速度仍小于加热器的供热速度,所以恒温水浴的温度T将会继续有一个小幅度的升高,至T>T'。当加热器中所散发的余热的减少,恒温水浴的散热速度将会等于加热器的供热速度,此时水温不再上升。随着加热器中的余热的不断减少,恒温水浴的散热速度将大于加热器的供热速度,也就必然导致水浴的温度T降低。当刚好满足T
从上面的讨论我们可以发现,恒温水浴的实际温度T一定会在我们所设定的温度T'的上下的一个小范围内波动。显然,这个温度波动范围越小,则表明恒温水浴的恒温性能就越好,本实验的目的之一就是测定这个波动范围(即恒温水浴的灵敏度曲线,如图1-1)。习惯上,
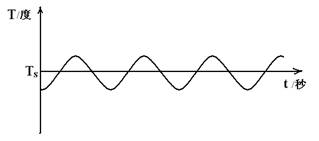
图1-1 控温灵敏度曲线
我们一般用
S /℃ = ±(T2-T1) / 2
表示恒温水浴的灵敏度,上式中T1、T2分别表示恒温过程中水浴的最高温度和最低温度。
四、实验内容与步骤
1. 检查和熟悉恒温水浴装置各部分的连接状态:数字式温控器(含电子继电器、温度计)、加热器(含温度设定装置)、数显贝克曼温度计。
2. 将数显贝克曼温度计设定为温差显示方式(为什么不用温度显示方式?)。将数字式温控器和数显贝克曼温度计的探头置于水浴中的同一区域,测定数字式温控器和数显贝克曼温度计测量温度时的相对偏差。相对偏差=(贝克曼温度计的温差+贝克曼温度计的基温)-数字式温控器的温度。
3. 接通电源,并打开各装置的电源开关,使用低功率加热和高速搅拌。
4. 通过数字式温控器的设定水浴温度(比实验开始时水温高5℃左右),并设置数字式温控器的回差为0.1。记录水浴的实际设定温度Ts =数字式温控器的温度±相对偏差。
5. 将恒温水浴通电15分钟,使恒温水浴稳定下来。接下来尝试每隔数秒钟记录一次温差读数及记录读数的时间。在读数过程中一定要认真观察,除按一定的时间间隔读出温差外,对水浴的最高温差(对应于水浴的最高温度)、最低温差(对应于水浴的最低温度)以及出现这两个温差的时间一定要同时记录下来。至于记录温差的时间间隔有学生自行设定,但要满足如下作图要求:为使作出的恒温水浴的灵敏度曲线较为光滑,除槽温波动过程中的最高点、最低点外,每一次槽温升高的过程中要求各记录2组数据点,而每一次降温过程中要各记录3组数据点。
6. 连续读数,记录温差、时间变化值,直到获得了5组连续的槽温波动-时间数据为止。
7. 停止实验,先关闭各装置的电源开关,再拔下插头,后并取出、擦干数字式温控器、数显贝克曼温度计的探头。
8. 打扫小组实验台面的卫生,抄录一份原始实验数据记录,交教师签字后离开实验室。
五、思考题
简要分析:①水浴的搅拌速度的快慢、②加热器功率的大小对恒温浴灵敏度的影响。
实验二 燃烧热的测定
一、实验目的
1.明确燃烧热的定义,了解恒压燃烧热和恒容燃烧热的差别及相互关系。
2.了解量热法的基本原理和一般的测试方法。
3.通过萘的燃烧热的测定,了解氧弹式量热计的原理、构造及使用方法。
4.学会雷诺图解法校正温度改变值。
二、实验主要设备及使用要求
氧弹计、压片器


图2-1氧弹量热计示意图 图2-2氧弹的构造
1.外筒,通过搅拌器搅拌形成恒温环境;2.绝热定位圈; 1.氧弹头,既是充气头,又是放气头;
3.氧弹;4.水桶,用以盛装量热介质;5.电极; 2.氧弹盖;3.电极;4.引火丝;
6.水桶搅拌器;7.温度传感器探头;8.外筒搅拌器 5.燃烧杯;6.燃烧挡板;7.卡套;8.氧弹体
三、实验原理、方法和手段
1.燃烧与热量
根据热化学的定义,燃烧热是指1mol物质完全燃烧时所放出的热量。在恒容条件下测得的燃烧热称为恒容燃烧热(Qv),在恒压条件下测得的燃烧热称为恒压燃烧热(Qp)。
△n为反应前后反应物和生成物中气体的物质的量之差,R为气体常数,T为反应时的热力学温度。由上式可知,若测得某物质的Qp或Qv中的任何一个,即可计算出另一个数据。在热化学中,化学反应的热效应(包括燃烧热)通常是用Qp来表示的。本实验测定的是Qv,所以需要通过上式进行计算。
2.氧弹量热计
直接测定Qv或Qp的实验方法称为量热法。通常能直接测定的热效应,除了燃烧热外,还有中和热、溶解热、稀释热和物质的比热等。这些热效应的数据,常用于其它热力学量的计算,并对化工设计和实际生产都有重要意义。
图2-1是氧弹量热计的结构示意图,这种量热计常用于物质燃烧热的测定。在量热计的外面有一个套壳,套壳有些是恒温的,有些是绝热的。本实验采用外壳等温式量热计。氧弹是一个具有良好密封性能、耐高压、抗腐蚀的不锈钢容器,其结构如图2-2所示。
原理:能量守恒定律
Q放=Q吸
-W样.QV,样/M样-L铁丝.QL,铁丝=(W水C水+C计)△T
QL,铁丝=-2.9J/cm
3.雷诺温度校正图
实际上量热计与周环境的热交换无法完全避免,它对温差的影响可用雷诺(Renolds)温度校正图校正。
使水温低于室温1℃左右,按实验步骤操作测燃烧热所得的一系列水温和时间关系作图.(p.45)。用微机控制时,初期温度:11个,主期温度:22个,末期温度:11个(每隔30秒自动记录一个数据)
说明:雷诺温度校正图2-3和2-4,AA’环境与搅拌引起的温升应扣除;CC’由室温升高到最高点,这一段时间内,量热计向环境的热漏造成的温度降低,应计算在内;所以温升为AC。(具体校正过程参阅实验教材)
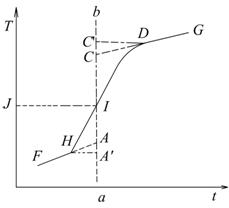
图2-3绝热较差时的雷诺校正图 图2-4绝热良好时的雷诺校正图
本实验所用温度计为贝克曼数显温度计,WHR-15微机型氧弹式热量计。
四、实验内容与步骤
1.热量计热容量的测定
所用试剂和材料
苯甲酸:已知恒压燃烧热焓,作基准物
萘:试样,作待测物
点火丝:铁丝,其燃烧值为2.9J/cm
氧气:装在专用钢瓶内压力可达150kg/cm2(15Mpa),禁火禁油。
2.热量计热容量的测定
(1)样品制作:用电子台称粗称约1克(不要超过1g)苯甲酸(W样),用压片机压成圆柱
型,取出来后再用电子天平准确称量至万分之一(0.0001)克。
(2)装样:拧开氧弹盖,将弹头放到弹头架上,用干毛巾将氧弹内外壁擦干净,在氧弹内装
入10ml水,将苯甲酸小心平放入弹头装样的坩埚内。剪取12cm长的点火丝,压制成
V字型,两端用螺丝固定在两个电极上,其中段贴压在苯甲酸上。点火丝切勿接触坩埚。
小心安装弹头,拧紧氧弹上盖,用万用表欧姆档,测量氧弹两电极电阻,电路畅通,电
阻不大于20欧姆。
(3)充氧:小心平稳地把单头氧弹立放在已连接好减压阀和氧气瓶的立式充氧器底板上,旋
开氧气钢瓶,减压阀压力调整为2Mpa,将单头氧弹进气咀对准充氧器的出气咀,手持
操纵手柄,轻轻迅速地往下压,30秒钟即可充满氧弹,(压力达到实验的要求2Mpa),
立即将充氧手柄抬起来,完成充氧操作。
(4)装弹:将充好氧气的氧弹对准位置放入HR-15氧弹式热量计的量热容器(内筒)中,连
接好点火电源引线,用1000ml容量瓶,(调节内筒的水温使之低于外筒的水温0.7—1℃)
准确地加入3000ml自来水,加完后注意观察氧弹是否漏气,盖好热量计上盖。
(5)恒温:将测温探头放入外筒,开启HR-15B热量计多功能控制箱(开关在箱后面),观
察并记录外筒温度。3-5分钟后将测温探头插入内筒,启动搅拌开关,(测温探头和搅
拌器均不得接触氧弹及内筒!!),观察内筒温度。
(6)开启计算机,进入Windows98视窗操作系统,用鼠标双击热量计软件快捷方式图标,按
提示进行人机对话操作。
A.输入式样重量(g):(输入不能出错,若出错则退出再重新进入系统)
B.试样编号:1
C.发热量(J/g):(输入标样苯甲酸的发热量)
D.使用公式:奔特公式
E.实验内容:热容量。
设置好以上几项后,点击“开始实验”,进行实验,若联机正确,则多功能控制箱数显数字消失,计算机软件界面中温度显示区显示内桶温度,观察和用记录本记录好电脑显示的初期、主期、末期温度、仪器热容数据。
3.萘的燃烧热的测定(样品不同,操作步骤和前面基本一样)
(1)样品制作:用电子台称粗称约0.6克(不要超过0.6克)萘(W萘),用压片机压成圆柱
型,取出来后再用电子天平准确称量至万分之一(0.0001)克。
(2)装样:拧开氧弹盖,用干毛巾将氧弹内外壁擦干净,在氧弹内装入10ml水,将苯甲酸
小心平放入弹头装样的坩埚内。剪取12cm长的点火丝,压制成V字型,两端用螺丝固
定在两个电极上,其中段贴压在萘上。点火丝切勿接触坩埚。拧紧氧弹上盖,用万用表
欧姆档,测量氧弹两电极电阻,电路畅通,电阻不大于20欧姆。
(3)充氧:小心平稳地把单头氧弹立放在已连接好减压阀和氧气瓶的立式充氧器底板上,减
压阀压力调整为2Mpa,将单头氧弹进气咀对准充氧器的出气咀,手持操纵手柄,轻轻
迅速地往下压,30秒钟即可充满氧弹,(压力达到实验的要求2Mpa),立即将充氧手柄
抬起来,完成充氧操作。
(4)装弹:将充好氧气的氧弹对准位置放入HR-15氧弹式热量计的量热容器(内筒)中,连
接好电源引线,用1000ml容量瓶(调节内筒的水温使之低于外筒的水温0.7—1℃),
准确地加入3000ml自来水,盖好热量计上盖。
(5)恒温:将测温探头放入外筒,开启HR-15B热量计多功能控制箱(开关在箱后面),观
察并记录外筒温度。3—5分钟后将测温探头插入内筒,启动搅拌开关,(测温探头和搅
拌器均不得接触氧弹及内筒!!),观察内筒温度。
(6)开启计算机,进入Windows98视窗操作系统,用鼠标双击热量计软件快捷方式图标,按
提示进行人机对话操作。
A.输入式样重量(g):(输入不能出错,若出错则退出再重新进入系统)
B.试样编号:2
C.热容量(J/℃):(输入第一步测得的热容量数据或计算自动输入)
D.使用公式:奔特公式
E.实验内容:发热量。
设置好以上几项后,点击“开始实验”,若联机正确,则多功能控制箱数显数字消失,计算机软件界面中温度显示区显示内桶温度,观察和用记录本记录好电脑显示的初期、主期、末期温度、燃烧热数据。原始数据记录本交给老师签字。
实验结束后,将实验数据转存于A盘(软盘)。用打印机打印出燃烧热测试报告单(贴到实验报告上)。退出实验系统,关好计算机及复原仪器。
五、思考题
1.在氧弹里加10mL蒸馏水起什么作用?
2.试根据实验所得的雷诺校正图,解释体系和环境热交换的情况。
3.水桶中的水温为什么要选择比外筒水温低?低多少合适?为什么?
4.如果要提高实验的准确度应从哪几个方面考虑。
5.如何用萘的燃烧热数据来计算萘的标准生成热。
C10H8(s)+12O2(g)→10CO2(g)+4H2O(l)
六、注意事项及其它说明
1.为保证测试温度,本仪器在使用前必须开机预热半小时
2.内筒中加一定体积的水后若有气泡逸出,说明氧弹漏气,设法排除。
3.搅拌时不得有摩擦声。
4.燃烧样品萘时,内筒水要更换且需重新调温
5.每次实验结束,要小心地拿出测温探头,再打开量热计的上盖,以免损坏测温探头。把所用仪器如氧弹、坩锅、内桶等清洗干净并放回原处。
6.顺时针旋转氧气钢瓶阀杆,是关好钢瓶。反时针旋转减压阀杆,是关好减压阀。要特别注意安全。开时先开氧气钢瓶,再开减压阀;关时先关钢瓶,再关减压阀。实验过程中,不用频繁开关钢瓶和减压阀。实验结束时,才要关好钢瓶和减压阀。(为什么?)
7.实验过程中不要做与本实验无关的事,不要动与本实验无关的仪器。
实验三 碳酸钙分解反应热的测定
一、实验目的
1. 测定650-850℃范围内,5-7点温度下的碳酸钙分解压,从而计算出在该温度范围内碳酸钙分解反应热。
2.了解一般的真空技术、压力、温度的测量方法。
二、实验主要设备及使用要求
实验装置:(如图3-1)
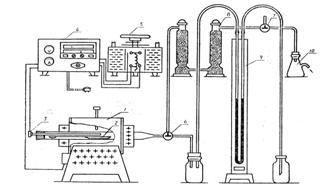
图3-1碳酸钙分解实验装置图
1.高温燃烧定碳炉;2.瓷舟;3.热电偶;4.温度控制器;5.调压器;
6、7 三通阀门;8.干燥塔;9.压力计;10.安全瓶
三、实验原理、方法和手段
CaCO3较高温度时,按下式分解,并吸取一定的热量
CaCO3→CaO(S)+CO2(g)
当温度不变时,反应将达到平衡,因此其平衡常数Kp=Pco2,其中Pco2是反应达到平衡时CO2的压力。
由等压方程式dlnKtp/dT=∆H/RT2积分可得
lnKtp==-∆H/RT+∆S/R
2.303Lg Pco2,=-∆H/RT+∆S/R
令-∆H/R2.303=A, ∆S/R2.303=B则原式为
lg Pco2=A/T+B
∆H—碳酸钙分解反应热; ∆S—体系的熵变量
因此在650—850℃范围内,将∆H和∆S分别看作常量,这样以1/T为横轴,以lg Pco2为纵轴,在坐标纸上作图,可得到一直线,通过求直线的斜率和截距即可求出∆H和∆S。
四、实验内容与步骤
1.装置碳酸钙粉末,用小勾勾出炉中的瓷舟,装满碳酸钙粉末,送入电炉的中心部位,塞
紧胶塞。
2.抽真空,先将与电炉相连的三通阀门置于三通的位置,与真空泵相连三通阀门置于与外
界隔断而呈直通的位置,再启动真空吧泵,将系统抽真空,待到压力计的液面高度不再
变化后,停止抽真空,欲停止抽真空时,应先将与真空泵相连三通旋转900,让安全瓶
进入空气后,再停止电机的转动。
3.设定温度。通电,设定温度控制器的温度为650℃,同时调节电炉的加热电压为70V,待到温度升到500℃时,可及时降低加热电压,减小电炉的功率,以免余热过大,造成温度不准。
4.二次抽真空。如果当温度为500℃时,压力计的差值与开始时的数值相差较大,应进行二次抽真空,方法如下:首先启动真空泵,然后按逆时针方向旋转真空泵相连三通旋转900,使该三通呈直通,二次抽真空时间不可过长,以免二氧化碳损失过多,大约1—2分钟,即可。二次抽真空停止时,先将与电炉相连的三通阀门置于直通的位置,再将与真空泵相连三通置于三通的位置。再停止电机的转动。
5.测量分解压及分解温度。当温度升到650℃时,电炉温度控制器会自动切断电源,使温度维持恒定不变,观察压力计液面波动情况,当变化小时,即可读出压力计的液面高度,同时记录温度的数值,然后设定温度为下一个测量点,测到850℃结束。
6.停止实验后切断电源,注意此时的瓷舟不可取出、触摸,以免伤人。
五、思考题
1.在实验中,温度是通过什么元件测量出来的?测量原理是什么?
2.停止抽真空时,应注意哪些问题?为什么?
实验四 溶解热的测定
一、实验目的
1.掌握量热技术及电热补偿法测定热效应的基本原理。
2.用电热补偿法测定KNO3在不同浓度水溶液中的积分溶解热。
3.用作图法求KNO3在水中的微分冲淡热、积分冲淡热和微分溶解热。
二、实验主要设备及使用要求
实验装置1套(包括杜瓦瓶、搅拌器、加热器、测温部件、加样漏斗;直流稳压电源;电子分析天平;直流毫安表;直流伏特计;)
三、实验原理、方法和手段
1.在热化学中,关于溶解过程的热效应,引进下列几个基本概念。
溶解热 在恒温恒压下,n2摩尔溶质溶于n1摩尔溶剂(或溶于某浓度的溶液)中产生的热效应,用Q表示,溶解热可分为积分(或称变浓)溶解热和微分(或称定浓)溶解热。
积分溶解热 在恒温恒压下,一摩尔溶质溶于n0摩尔溶剂中产生的热效应,用Qs表示。
微分溶解热 在恒温恒压下,一摩尔溶质溶于某一确定浓度的无限量的溶液中产生的热效应,以表示,简写为。
冲淡热 在恒温恒压下,一摩尔溶剂加到某浓度的溶液中使之冲淡所产生的热效应。冲淡热也可分为积分(或变浓)冲淡热和微分(或定浓)冲淡热两种。
积分冲淡热 在恒温恒压下,把原含一摩尔溶质及n01摩尔溶剂的溶液冲淡到含溶剂为n02时的热效应,亦即为某两浓度溶液的积分溶解热之差,以Qd表示。
微分冲淡热 在恒温恒压下,一摩尔溶剂加入某一确定浓度的无限量的溶液中产生的热效应,以表示,简写为。
2.积分溶解热(QS)可由实验直接测定,其它三种热效应则通过QS—n0曲线求得
设纯溶剂和纯溶质的摩尔焓分别为Hm(1)和Hm(2),当溶质溶解于溶剂变成溶液后,在溶液中溶剂和溶质的偏摩尔焓分别为H1,m和H2,m,对于由n1摩尔溶剂和n2摩尔溶质组成的体系,在溶解前体系总焓为H。
H=n1Hm(1)+n2Hm(2) (1)
设溶液的焓为H′,
H′=n1H1,m+n2H2,m (2)
因此溶解过程热效应Q为
Q=ΔmixH=H-H=n1[H1。m–Hm(1)]+n2[H2,m–Hm(2)]
=n1ΔmixHm(1)+n2ΔmixHm(2) (3)
(3)式中,ΔmixHm(1)为微分冲淡热,ΔmixHm(2)为微分溶解热。根据上述定义,积分溶解热QS为
(4)
在恒压条件下,Q=ΔmixH,对Q进行全微分
(5)
上式在比值恒定下积分,得
(6)
全式以n2除之
(7)
因
(8)
则 (9)
将(8)、(9)代入(7)得:
(10)
对比(3)与(6)或(4)与(10)式,
以QS对n0作图,可得图1的曲线关系。在图1中,AF与BG分别为将一摩尔溶质溶于n01和n02摩尔溶剂时的积分溶解热QS,BE表示在含有一摩尔溶质的溶液中加入溶剂,使溶剂量由n01摩尔增加到n02摩尔过程的积分冲淡热Qd。
Qd=(QS)n02- (QS)n01=BG – EG (11)
图4-1中曲线A点的切线斜率等于该浓度溶液的微分冲淡热。
切线在纵轴上的截距等于该浓度的微分溶解热。
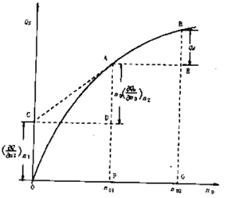
图4-1QS—n0关系图
由图4-1可见,欲求溶解过程的各种热效应,首先要测定各种浓度下的积分溶解热,然后作图计算。
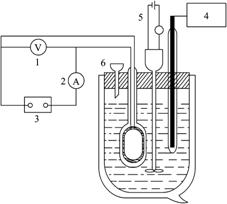
图4-2量热器及其电路图
1.直流伏特计;2.直流毫安表;3.直流稳压电源;4.测温部件;5.搅拌器;6.加样漏斗
3.本实验是采用绝热式测温量热计,它是一个包括量热器、搅拌器、电加热器和温度计等的量热系统,装置及电路图如图4-2所示,因本实验测定KNO3在水中的溶解热是一个吸热过程,可用电热补偿法,即先测定体系的起始温度T,溶解过程中体系温度随吸热反应进行而降低,再用电加热法使体系升温至起始温度,根据所消耗电能求出热效应Q。
Q=I2Rt=IUt
式中,I为通过电阻为R的电热器的电流强度(A);U为电阻丝两端所加电压(V);t为通电时间(s).这种方法称为电热补偿法。
本实验采用电热补偿法,测定KNO3在水溶液中的积分溶解热,并通过图解法求出其它三种热效应。
四、实验内容与步骤
1.将8个称量瓶编号,在台秤上称量,依次加入在研钵中研细的KNO3,其重量分别为2.5g、1.5g、2.5g、2.5g、3.5g、4g、4g和4.5g,再用分析天平称出准确数据,称量后将称量瓶放入放入干燥器中待用。
2.在台称上用杜瓦瓶直接称取200.0g蒸馏水,调好贝克曼温度计,按图2装好量热器。按图3连好线路(杜瓦瓶用前需干燥)。
3.经教师检查无误后接通电源,调节稳压电源,使加热器功率约为2.5W,保持电流稳定,开动搅拌器进行搅拌,当水温慢慢上升到比室温水高出1.5℃时读取准确温度,按下秒表开始计时,同时从加样漏斗处加入第一份样品,并将残留在漏斗上的少量KNO3全部掸入杜瓦瓶中,然后用塞子堵住加样口。记录电压和电流值,在实验过程中要一直搅拌液体,加入KNO3后,温度会很快下降,然后再慢慢上升,待上升至起始温度点时,记下时间(读准至秒,注意此时切勿把秒表按停),并立即加入第二份样品,按上述步骤继续测定,直至八份样品全部加完为止。
4.测定完毕后,切断电源,打开量热计,检查KNO3是否溶完,如未全溶,则必须重作;溶解完全,可将溶液倒入回收瓶中,把量热器等器皿洗净放回原处。
5.用分析天平称量已倒出KNO3样品的空称量瓶,求出各次加入KNO3的准确重量。
五、思考题
对本实验的装置你有何改进意见?
本实验的装置可从以下几个方面改进:(1)本实验用电热补偿法测量溶解热时,整个实验过程要注意电热功率的检测准确,但由于实验过程中电压波动的关系,很难得到一个准确值。如果实验装置使用计算机控制技术,采用传感器收集数据,使整个实验自动化完成,则可以提高实验的准确度。(2)本实验采用绝热式量热计,但在加样的过程中存在热损失,不可能做到完全的绝热,可考虑采用合适的加样器,包含在绝热系统之内。(3)本实验采用杜瓦瓶作为反应装置,可视性不好,可以考虑用透明的绝热玻璃装置,增强反应的透明度,更容易观察硝酸钾的溶解性。
六、注意事项及其它说明
1.实验过程中要求I、V值恒定,故应随时注意调节。
2.磁子的搅拌速度是实验成败的关键,磁子的转速不可过快。
3.实验过程中切勿把秒表按停读数,直到最后方可停表。
4.固体KNO3易吸水,故称量和加样动作应迅速。固体KNO3在实验前务必研磨成粉状,并在110℃烘干。
5.量热器绝热性能与盖上各孔隙密封程度有关,实验过程中要注意盖好,减少热损失。
实验五 纯液体饱和蒸气压的测定
一、实验目的
1.用平衡管测定不同温度下液体的饱和蒸气压。
2.了解纯液体的饱和蒸气压与温度的关系。克劳修斯-克拉贝龙(Clausius-Clapeyron)方程式的意义,并学会由图解法求其平均摩尔气化热和正常沸点。
二、实验主要设备及使用要求
饱和蒸气压装置
实验装置:
图5-2纯液体饱和蒸气压测定装置图
精密数字压力计表的使用:
(1) 预热:将开关置于标有“ON”的位置,通电预热30min后,方可进行实验测定,否则将影响实验精度。
(2) 调零:按“采零”键,调节零点的读数为“0.00”,重复2~3次。
(3) 关机:关机前,应使测量体系与大气相连通后方可关机。
(4) 压力计是以实验室的大气压作为“0”基准点进行测压。例如:实验室的大气压为99.65KPa,仪器在完全连通实验室大气压的状态下“采零”,实验过程中,如果压力计的示数为-20KPa,则体系的真实压力比实验室的大气压小20 KPa,为79.65KPa。
恒温水浴调节:温度恒定5min后才能测定。
三、实验原理、方法和手段
在通常温度下(距离临界温度较远时),密闭真空容器中,纯液体与其蒸气达平衡时的蒸气压力称为该温度下液体的饱和蒸气压,简称为蒸气压。蒸发一摩尔液体所吸收的热量称为该温度下液体的摩尔气化热。
 液体的蒸气压随温度而变化,温度升高时,蒸气压增大;温度降低时,蒸气压降低,这主要与分子的动能有关。当蒸气压等于外界压力时,液体便沸腾,此时的温度称为沸点,外压不同时,液体沸点将相应改变,当外压为101.325kPa时,液体的沸点称为该液体的正常沸点。
液体的蒸气压随温度而变化,温度升高时,蒸气压增大;温度降低时,蒸气压降低,这主要与分子的动能有关。当蒸气压等于外界压力时,液体便沸腾,此时的温度称为沸点,外压不同时,液体沸点将相应改变,当外压为101.325kPa时,液体的沸点称为该液体的正常沸点。
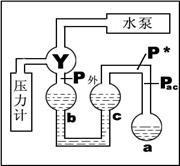
1、 饱和蒸气压的测定(见图5-1):
(1)抽气减压,使Pac=P*;
(关闭通气阀1,打开通气阀2及抽气阀,
用水泵抽气减压)
(2)调节b、c两液面相平,测定Pb,则
Pb=P*。(关闭通气阀2,缓慢打开通气阀1)
2、 平均摩尔气化热ΔvHm的测定: 图5-1 实验原理图
根据克劳修斯—克拉帕龙方程,有:
式中,R为摩尔气体常数;T为热力学温度;ΔvapHm为在温度T时纯液体的摩尔气化热。
假定ΔvapHm与温度无关,或因温度范围较小,ΔvapHm可以近似作为常数,积分上式,得:
其中:C是积分常数,以 lnp 对1/T作图,应为一直线,直线的斜率为-△vapHm/R,由斜率可求算液体的ΔvapHm。
测定液体饱和蒸气压的方法很多。本实验采用静态法,是指在某一温度下,直接测量饱和蒸气压,此法一般适用于蒸气压比较大的液体。
四、实验内容与步骤
1.装样:吹热A球(排空气)→将蒸馏水放入等压计B、C间的U形管中→自然冷却→蒸馏水吸入A球→重复几次→蒸馏水装入A球的2/3。
2.检漏:
接通冷凝水→打开平衡阀1、平衡阀2及抽气阀→调节恒温槽温度为实验温度t1=50℃并恒温10min→压力计“采零”(按“采零”键)→关闭平衡阀1→打开抽气水泵→抽气减压→压力计读数为53~67 KPa→关闭平衡阀2→观察是否漏气。
3.测定:
同一温度t下的测定:
① 如果不漏气则可直接进行以下步骤:(接2的检漏步骤)→打开平衡阀2→抽气减压→U型管等位计中b、C间有气泡形成并逐渐排出→有连续气泡排出→关闭平衡阀2(如果液体暴沸,稍微打开平衡阀1使沸腾缓和后再关闭平衡阀1)→缓慢沸腾3~4min →缓慢打开平衡阀1使b、c液面相平→读出压力计的压力值。
② 关闭平衡阀1,打开平衡阀2→U型管等位计中b、C间有气泡形成并逐渐排出→有连续气泡排出→关闭平衡阀2(如果液体暴沸,稍微打开平衡阀1使沸腾缓和后再关闭平衡阀1)→缓慢沸腾3~4min →缓慢打开平衡阀1,调节b、c液面相平→读出压力计的压力值。(2次)
不同温度下的测定:
(接上步骤)缓慢打开平衡阀1、平衡阀2及抽气阀→压力计读数显示为“0.00” KPa→关闭水泵→调节恒温槽温度为实验温度tˊ,使tˊ=55、60、65、70℃,分别重复(1)实验步骤。
五、思考题
1.压力与温度的测量都有随机误差,试导出的误差传递表达式。
2.用此装置可以很方便地研究各种液体,如苯、二氯乙烯、四氯化碳、水、正丙醇、异丙醇、丙酮和乙醇等,这些液体中很多是易燃地,在加热时应该注意什么问题?
六、注意事项及其它说明
1.实验前必须严格捡漏,否则抽气时达不到本实验要求的真空度。
2.注意控温,测量中,恒温水浴的回差控制应在最小值,等压计必须垂直,且全部放置于恒温水浴中的液面下,否则其温度与水浴温度不同。
3.必须排净U形管空中全部空气,使A球液面上空只含液体的蒸气分子。
4.实验过程中严防空气倒灌,否则实验重新操作。
5.防止液体暴沸。
6.水泵操作要缓慢,关停水泵前,要先使测量系统与大气相连通,防止水泵中的水倒灌。
实验六 双液系气—液平衡相图
一、实验目的
1.绘制在p下环已烷-乙醇双液系的气----液平衡图,了解相图和相率的基本概念。
2.掌握测定双组分液系的沸点的方法,找出恒沸点混合物的组成和最低恒沸点。
3.掌握用折光率确定二元液体组成的方法
4.掌握阿贝折射仪的测量原理及使用方法。
二、实验主要设备及使用要求
沸点仪(见图6-2)1套;超级恒温水浴;阿贝折光仪;
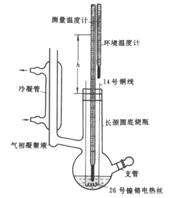
1.阿贝折光仪的使用及说明
根据折射定律,波长一定的单色光在确定的外界条件下(温度、压力等),从一种介质A进入另一种介质B时,入射角α和折射角β的正弦之比与两种介质的折射率N与n成反比:
sinα/sinβ=n/N (1)
当介质A为真空时,N=1,n为介质B的绝对折射率,则有
n = sinα/sinβ (2)
如果介质A为空气,N空气=1.00027(绝对折射率),则:
sinα/sinβ=n/N空气=n/1.00027 =n´ (3)
n´为介质B的相对折射率。n与n´数值相差很小,常以n代替n´。但进行精密测定时,应加以校正。n与物质结构、光的波长、温度及压力等因素有关:
(1)通常大气压的变化影响不明显;
(2)使用单色光要比白光时测得的n值更为精确,因此,常用钠光(D)(λ=28.9nm)作光源;
(3)温度可用恒温水浴槽与折光仪间循环恒温水来维持恒定温度。折射率表示为nD20,即以钠光灯为
光源,20℃时所测定的n值。
把t℃时测得的折光率校正
到20℃时的折光率:
n=nt+ 0.00045×(t-20℃) 图6-3 阿贝折光仪
2.阿贝折光仪的操作方法
(1)用擦镜纸将镜面擦干,取样管垂直向下将样品滴加在镜面上,注意不要有气泡,然后将上棱镜合上,关上旋钮。
(2)打开遮光板,合上反射镜。
(3)轻轻旋转目镜,使视野最清晰。
(4)旋转刻度调节手轮,使目镜中出现明暗面(中间有色散面)
(5)旋转色散调节手轮,使目镜中色散面消失,出现半明半暗面。
(6)再旋转刻度调节手轮,使分界线处在十字相交点。在下标尺上读取样品的折光率。
三、实验原理、方法和手段
液体的沸点是指液体的饱和蒸汽压和外压相等时的温度。在一定外压下,纯液体的沸点有确定的值。但对于完全互溶的双液系,沸点不仅与外压有关,而且还与双液系的组成有关。
常温下,两种液态物质以任意比例相互溶解所组成的体系称为完全互溶双液系。在恒定压力下,表示溶液沸点与组成关系的相图称为沸点—组成图,即为T-x相图。完全互溶双液系的T-x图可分为三类(见图6-1):
(1)理想双液系,溶液沸点介于两纯物质沸点之间如图(a);
(2)各组分对拉乌尔定律发生正偏差,溶液具有最低恒沸点(图中最低点)如图(b);
(3)各组分对拉乌尔定律发生负偏差,溶液具有最高恒沸点(图中最高点)如图(c);
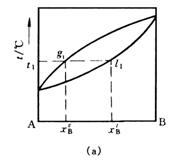
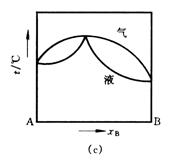
图6-1 完全互溶双液系的T-x图
绘制双液系的T-x图时,需要同时测定气液平衡时溶液的沸点及气相组成、液相组成数据。例如图(a)中,与沸点t1对应的气相组成是气相线上g1点对应的,液相组成是液相线上l1点对应的。实验测定整个浓度范围内不同组成溶液的气液相平衡组成和沸点后,即可绘出T-x图。
本实验采用回流冷凝的方法绘制环己烷-乙醇体系的T-x图。其方法是用Abbe折射仪测定不同组分的体系在沸点时气液两相的折光率。在折光率-组成图(标准曲线)找出未知浓度溶液的折光率,就可从曲线上查出相对应的组成
四、实验内容与步骤
1.沸点仪的安装(如右图所示);调节恒温槽温度30℃,通恒温水于折光仪中。打开折光仪,预热。
2.绘制标准曲线(因操作时间关系,由教师给出标准曲线数据)
3.以5ml无水乙醇润洗沸点仪,重复操作一次。
4.加入无水乙醇20ml,盖好瓶塞,使电热丝浸入液体中,温度计水银球与液面接触。开冷凝水。
5.选择合适电压(8—10V)接通电热丝至沸腾温度,待温度恒定后,读下该温度值。
6.停止加热,将取样管自冷凝管上端插入冷凝液收集小槽中,取气相冷凝液样,迅速用阿贝折射仪测其折光率。
7.取液相液样,用阿贝折射仪测其折光率。
8.在沸点仪中加入环已烷0.5ml,重复上述5、6、7操作,记录气相及液相折光率数据。
9.继续分别加入环已烷1.0ml、1.5ml、2.0ml、4.0ml、14ml。重复上述5、6、7操作,记录气相及液相折光率数据,将溶液倒入回收瓶中,洗净沸点仪,干燥。
10.加入环已烷25ml,重复上述5、6、7操作,继续分别加入无水乙醇0.1ml、0.2ml、0.3ml、0.4ml、1.0ml、5.0ml。重复上述5、6、7操作,记录每次操作的气相及液相折光率数据。
11.根据环已烷-乙醇的标准溶液的折射率,将上述数据转换成环已烷的摩尔分数,绘制相图。
实验完毕后,关闭冷凝水,关闭电源,清洗仪器,整理实验台。
五、思考题
1.在测定沸点时,溶液过热或出现分馏现象,将使绘出的相图图形发生变化?
2.为什么工业上常生产95%酒精?只用精馏含水酒精的方法是否可能获得无水酒精?
3.讨论本实验的主要误差来源。
4.在连续测定法实验中,样品的加入量应十分精确吗?
六、注意事项及其它说明
1.在测定纯液体样品时,沸点仪必须是干燥的。
2.在整个实验中,取样管必须是干燥的。
3.本实验的超级恒温槽的温度必须调至30℃(因为本实验环已烷-乙醇的标准溶液的折射率是在30℃测定)
4. 将样品取样至阿贝折射仪测定时,取样管应该垂直向下。
5. 在使用阿贝折射仪读取数据时,特别要注意在气相冷凝液样与液相液样之间一定要用擦镜纸将镜面擦干。
6. 注意线路的连接,加热时,应缓慢将变压器调至合适电压(8-10V)。如电热丝未接通,则关闭电源,首先检查线路,然后检查电热丝的接触情况,进行适当调整。
实验七 二组分金属相图的绘制
一、实验目的
1.用热分析法(步冷曲线法)测绘Pb—Sn二组分金属相图。
2.了解固液相图的特点,进一步学习和巩固相律等有关知识。
3.掌握热电偶测量温度的基本原理。
二、实验主要设备及使用要求
JXL-2型金属相图炉;微电脑温度控制仪;铂电阻温度计

图7-1步冷曲线测定装置示意图
1.相图炉;2.样品管;3.铂电极;4.电压表;5.电压钮;6.温度控制仪;7.温度显示屏;8.温度设置;9.定时器;l0.复位器
三、实验原理、方法和手段
较为简单的二组分金属相图主要有三种:一种是液相完全互溶;凝固后,固相也能完全互溶成固体混合物的系统,最典型的为Cu-Ni系统;另一种是液相完全互溶而固相完全不互溶的系统,最典型的是Bi-Cd系统;还有一种是液相完全互溶,而固相是部分互溶的系统,如Pb-Sn系统。
热分析法(步冷曲线法)是绘制相图的基本方法之一。它是利用金属及合金在加热和冷却过程中发生相变时,潜热的释出或吸收及热容的突变,来得到金属或合金中相转变温度的方法。
通常的做法是先将金属或合金全部熔化,然后让其在一定的环境中自行冷却,并在记录仪上自动画出(或人工画出)温度随时间变化的步冷曲线(见图7-2)。
当熔融的系统均匀冷却时,如果系统不发生相变,则系统的温度随时间的变化是均匀的,冷却速率较快(如图中ab线段);若在冷却过程中发生了相变.,由于在相变过程中伴随着放热效应,所以系统的温度随时间变化的速率发生改变,系统的冷却速率减慢,步冷曲线上出现转折(如图中b点)。当熔液继续冷却到某一点时(如图中c点),此时熔液系统以低共熔混合物的固体析出。在低共熔混合物全部凝固以前,系统温度保持不变,因此步冷曲线上出现水平线段(如图中cd线段);当熔液完全凝固后;温度才迅速下降(如图中de线段)。
由此可知,对组成一定的二组分低共熔混合物系统,可以根据它的步冷曲线得出有固体析出的温度和低共熔点温度。根据一系列组成不同系统的步冷曲线的各转折点,即可画出二组分系统的相图(温度-组成图)。不同组成熔液的步冷曲线对应的相图如图7-3所示。
用热分析法(步冷曲线法)绘制相图时,被测系统必须时时处于或接近相平衡状态,因此冷却速率要足够慢才能得到较好的结果。

图7-2 根据步冷曲线绘制相图 图7-3有过冷现象时的步冷曲线
四、实验内容与步骤
1.配制样品
在六个宽肩硬质玻璃管中,分别称重加入纯铅100,80,60,38.1,20,0 g,再分别加入已称重的纯锡0,20,40,61.9,80,100 g。样品上分别加少量石墨粉。
2.测定样品
将装有样品的试管放入相图炉内,把铂电极温度计插入样品管中(使其顶部离样品管底约1cm处)。接通电源,根据不同组成设置温度(见附表),使样品加热熔融。
炉温控制在以样品全部熔化后再升高50℃为宜。调节加热功率和冷却风速控制电炉的冷却速率,通常为每分钟下降6-8℃。每隔30s读取一次温度数值,直至三相共存温度以下约50℃。
五、思考题
1.对于不同成分的混合物的步冷曲线,其水平段有什么不同?为什么?
2.步冷曲线各段的斜率以及水平段的长短与哪些因素有关?
3.根据实验结果讨论各步冷曲线的降温速率控制是否得当。
4.作相图还有哪些方法?
六、注意事项及其它说明
1.加热样品时,注意控制温度,防止温度过热或过低。温度过高则样品易氧化变质,步冷曲线转折点测不准;温度过低或加热时间不够,则样品不能完全熔化,步冷曲线转折点测不出。
2.热电偶热端应插到样品中心部位,并在套管内注入少量的石蜡油,将热电偶浸入油中,以改善其导热情况。搅拌时要注意勿使热端离开样品,金属熔化后常使热电偶玻璃套管浮起,这些因素都会导致测温点变动,必须注意。
3.在测定一样品时,可将另一待测样品放入加热炉内预热,以便节约时间。混合物的体系有两个转折点,必须待第二个转折点测完后方可停止实验,否则须重新测定。
4.必须同时手工记录实验数据,以免意外情况造成数据丢失。
实验八 差热分析
一、实验目的
1.用差热—热重联用仪对CuSO4·5H2O进行差热及热重分析,并定性分析所测的差热—热重谱图;
2.掌握差热—热重分析原理,了解差热—热重分析仪的构造,学会操作方法。
二、实验主要设备及使用要求
DTG-60H差热—热重联用仪,仪器使用说明:
1.开机
打开电源开关,再打开计算机开关,然后打开主机等仪器的开关。双击“TA-60WS Collect”—File—ta—ch.1—DTG-60H。出现一兰色画面,说明仪器已联接完成。
2.放样品
按主机上“Open/Close”键,把炉子升起来,用镊子将装有参比物α—Al2O3(10mg左右)的铝坩埚轻轻放在炉子的左侧检测杆上,将另一同样质量的空铝坩埚轻轻放在炉子的右侧检测杆上,然后再按“Open/Close”键,把炉子降下来,之后按主机上“Display”键,仪器上显示样品的重量。因为左边放了样品,所以仪器上显示负值,按“Zero”键可以清零,清零之后再将炉子升起来,把右侧的坩埚拿出来,放入少量(10mg左右)CuSO4·5H2O晶体,样品颗粒尽量细小、均匀,并且最好平铺在坩埚底部。再将其重新放入炉内的右侧检测杆上,将炉子降下,按“Display”键,记下CuSO4·5H2O样品的重量。
3.设定程序
(1)Measure—Measure Parameters。设定升温速度,本实验为10℃/min,一般为2—20℃/min。最高温度可达1500℃,本实验为300℃,再选择是否需要保持温度,是否加保护气等,本实验均不需要。最后按确定。
(2)File Information。输入:1)批号;2)名称(可以用中文);3)样品重量;4)分子量;5)选择坩埚;6)选保护气;7)保护气流速(一般30ml/min);8)操作员。之后按确定。
4.全部确定后,按“Start”,炉子开始加热,计算机屏幕由兰色变成粉红色,加热结束后,屏幕又变成兰色。绿线—温度,红线—差热,兰线—热重。
三、实验原理、方法和手段
1.差热分析
差热分析是在程序控制温度下,测量试样与参比物(一种在测量温度范围内不发生任何热效应的物质)之间的温度差与温度关系的一种技术。
许多物质在加热或冷却过程中会发生熔化、凝固、晶型转变、分解、化合、吸附、脱附等物理化学变化。这些变化必将伴随体系焓的改变,因而产生热效应。其表现为该物质与外界环境之间有温度差。选择一种对热稳定的物质作为参比物,将其与样品一起置于可按设定速率升温的电炉中。分别记录参比物的温度以及样品与参比物间的温度 图8-1 时间温度变化图
差以温差对温度作图就可以得到一条差热分
析曲线,或称差热谱图,见图8-1。
如果参比物和被测物质的热容大致相同,而被测物质又无热效应,两者的温度基本相同,此时测到的是一条平滑的直线,该直线称为基线。一旦被测物质发生变化,因而产生了热效应,在差热分析曲线上就会有峰出现。热效应越大,峰的面积也就越大。在差热分析中通常还规定,峰顶向上的峰为放热峰,它表示被测物质的焓变小于零,其温度将高于参比物。相反,峰顶向下的峰为吸收峰,则表示试样的温度低于参比物。
差热曲线的峰形、出峰位置、峰面积等受被测物质的质量、热传导率、比热、粒度、填充的程度、周围气氛和升温速度等因素的影响。因此,要获得良好的再现性结果,对上述各点必须十分注意。一般而言,升温速度 图8-2 热重装置结构示意图
增大,达到峰值的温度向高温方向偏移;峰形变锐但峰的分辨率降低,两个相邻的峰,其中一个将会把另一个遮盖起来。
2.热重分析
当被测物质在加热过程中有升华、汽化、分解出气体或失去结晶水时,被测的物质质量就会发生变化。这时热重曲线就不是直线而是有所下降。通过分析热重曲线,就可以知道被测物质在多少度时产生变化,并且根据失重量,可以计算失去了多少物质,(如CuSO4·5H2O中的结晶水)。从热重曲线上我们就可以知道CuSO4·5H2O中的5个结晶水是分三步脱去的。图8-2为热重装置结构示意图。
四、实验内容与步骤
1.仔细阅读仪器操作说明书,在老师指导下,开启仪器(见图8-3)。
2.放样品。
3.设定程序。
4.加热(加热结束后,计算机自动将测得结果存入“TA60”)。
图8-3 实验装置示意图
5.数据分析:将“TA-60WS”窗口关上,双击TA-60WS—File
—Open—选择你所测得的文件后—打开。
1)换坐标轴。双击横坐标轴—Unit--选择Temp
(℃)—确定,就将横坐标轴由时间变为温度。双击纵
坐标轴(TGA)—Unit--选择TGA(%)—确定,(便于对不同重量的同一样品进行比较)
2)做切线找熔点:单击差热线,Analysis—Targent,从差热线的端点拉出两条线,分别放在要做切线的点两侧适当的位置,单击右侧“Analyze”出现切点,再单击“Analyze”出现温度值。
3)计算热重:单击热重线,Analysis—Weight Loss,从热重线的端点拉出两条线,分别放在有失重的线段的两端,单击“Analyze”出现失重的质量及失重百分含量。
4)多次做同一样品进行比较:File—Open—选择你所需要的文件—打开。Insert—Date File再打开另一张图,就可以把两张图放在一起比较了。
5)打印报告:File—Open—选择你所需要的文件—打开。View—Signed with Windows—空白处单击,将鼠标放在外框的边或角上,待出现←时,将框移动到所需大小的位置。
单击一条线—Insert—Title
单击一条线—Insert—File Information
单击一条线—Insert—Program
单击一条线—Insert—Legend
可以单击出现的文字,出现一个框,
将框移至图外,如果框内文字多,空白
处放不下,可以改变字体的大小,双击
框内字— Font— Change Font—选择
字号—确定—确定。
最后打印报告:File—Print,得图
8-4所示图谱。
图8-4 CuSO4·5H2O差热及热重分析图
五、思考题
1.DTA实验中如何选择参比物?常用的参比物有哪些?
2.差热曲线的形状与那些因素有关?影响差热分析结果的主要因素是什么?
3.DTA和简单热分析(步冷曲线法)有何异同?
4.根据无机化学知识和差热峰面积讨论五个结晶水与CuSO4结合的可能形式?
5.在什么情况下,升温过程与降温过程所得到的差热分析结果相同?在什么情况下,只能采用升温或降温的办法?
六、注意事项及其它说明
1.坩埚一定要清理干净,否则埚垢不仅影响导热,杂质在受热过程中也会发生物理化学变化,影响实验结果的准确性。
2. 样品必须研磨的很细,否则差热峰不明显;但也不宜太细。一般差热分析样品研磨到200目为宜。
3. 双笔记录仪的两支笔并非平行排列,为防二者在运动中相碰,制作仪器时,二者位置上下平移一段距离,称为笔距差。因此,在差热图上求转折温度时应加以校正。
实验九化学反应平衡常数和分配系数的测定
一、实验目的
测定KI+I2→KI3的平衡常数和[I2]H2O=[ I2]CCI4的分配系数
二、实验主要设备及使用要求
恒温水浴锅;碘素瓶;锥形瓶;滴定管
三、实验原理、方法和手段
在I2和KI的混合溶液中,存在下面的平衡关系即KI+I2→KI3
若在浓度不大的稀溶液中,则其反应的活度平衡常数近似等于浓度平衡常数,也就是
其中KC——化学反应平衡常数
CI2——平衡时I2的浓度
CI-3——平衡时KI3的浓度
CI-——平衡时KI的浓度
因此要测得它们的化学反应平衡常数,只要分别测出它们的浓度值,就可以。
1.CI2的测定
如果用0.01M的Na2S2O3直接滴定建立了化学平衡的反应液则由于I2+Na2S2O3= Na2S4O6+2NaI随着I2的消耗,将使化学平衡向左移动, KI3不断分解,以便建立新的平衡.所以最终测得的是平衡时CI2+CI-3的总和.,为解决这一问题,向溶液中加入CCI4,由I2在水中和CCI4的溶解度不同,将在溶液中建立如下的平衡关系,即:
[I2]H2O=[ I2]CCI4
首先测得该反应的分配系数,因为在一定温度的条件下该数值是一常数.只要分别提取达到平衡时的一定体积的水层和CCI4层进行分析,就可以求出K的大小。
K=[ I2]CCI4/[I2]H2O
其中 [ I2]CCI4----I2在CCI4层中的浓度,[I2]H2O_----I2在水层中的浓度
由于在混合液中加入了CCI4,所以只要分析一下CCI4层中I2的浓度由K值就可以求出I2在水层中的浓度,也就是CI2设为a。
2.CI-3的测定
提取一定体积的达到平衡的水层溶液,用0.01M的Na2S2O3直接滴定测出CI2+CI-3的总和.设为b则CI-3=b-a
3.CI-的测定
CI-的浓度应用初始浓度减去平衡时生成的KI3的浓度即CI-=0.05-(b-a)
Kc=(b-a)/[0.05-(b-a)]a
四、实验内容与步骤
1.调节恒温水浴锅,至250左右
2.按记录表中1、2号混合液的组成,分别配置1、2号混合液于相应的碘素瓶中,置恒温槽内1小时,在这期间应每隔5分钟取出一次振荡1分钟,使反应充分地达到平衡。
3.达到平衡后,应按记录表规定的体积进行取样分析,注意;在取CCI4层时,由于水层在上面,所以在移液管通过水层时,应不断用吸耳球向内鼓入空气,以防水进入移液管,影响测量精度。
4.分析水层时,应先加入2毫升的淀粉指示剂,用0.01M的Na2S2O3滴定到兰色消失为反应的终点,记下消耗的Na2S2O3的量。
5.分析CCI4层时,必须先加入10毫升左右的蒸馏水,使CCI4层的I2转移到水中去,因为I2只有在水中才能与Na2S2O3发生化学反应,所以必须充分地振荡,使CCI4层的I2转移到水中去,当水层呈现淡黄色时,加入2毫升的淀粉指示剂,呈现兰色再用Na2S2O3进行滴定,注意在滴定的过程中,一定是水层是兰色时,方可继续进行滴定,否则有可能超过等当点。一直到水层的颜色和CCI4层的颜色同时消失,才可停止,记下消耗的Na2S2O3的量。
6.实验结束后,将上层的水倒掉,CCI4层倒到回收瓶内。
五、思考题
1.在此实验中哪些溶液须准确配制,哪些不需要准确配制?
2.在I2和KI的混合溶液中,用Na2S2O3直接进行滴定分析,测得的是I2的浓度吗?为什么?
六、注意事项及其它说明
1.用标准Na2S2O3溶液滴定碘时,先要滴定至淡黄色再加淀粉溶液。
2.取CCl4层样品时勿使水层进入移液管中,为此用洗耳球使移液管尖鼓气情况下穿过水层插入CCl4层中取样。
3.在滴定CCl4层样品的I2时,应加入10ml0.1mol/L的KI水溶液以加快CCl4层中的I2完全提取到水层中,这样有利于Na2S2O3滴定的顺利进行。滴定时要充分摇荡,细心地滴至水层淀粉指示剂的蓝色消失,四氯化碳层不再现红色。
4.滴定后的和末用完的CCl4皆应倒人回收瓶中。
5.平衡常数和分配系数均与温度有关,因此本实验应严格控制温度。
实验十 分解反应平衡常数的测定
一、实验目的
1.测定氨基甲酸铵的分解压力,计算反应的平衡常数Kp;
2.熟悉用等压计测定平衡压力的方法;
3.根据氨基甲酸铵分解反应平衡常数计算有关热力学函数。
二、实验主要设备及使用要求
真空泵1台;低真空测压仪1台。
图10-1自制氨基甲酸铵反应装置示意图
图10-2静态平衡压力法测定压力装置示意图
三、实验原理、方法和手段
氨基甲酸铵是合成尿素的中间产物,为白色固体,很不稳定,其分解反应式为:
NH2COONH4(s)
 2NH3(g)+CO2(g)
2NH3(g)+CO2(g)
该反应为复相反应,在封闭体系中很容易达到平衡,在常压下其平衡常数可近似表示为:
(1)
式中,pNH 3、pCO 2分别表示反应温度下NH3和CO2平衡时的分压;在压力不大时,气体的逸度近似为1,且纯固态物质的活度为1,体系的总压p=pNH 3+pCO。从化学反应计量方程式可知:
(2)
将式(2)代入式(1)得:
(3)
因此,当体系达平衡后,测量其总压p,即可计算出平衡常数
温度对平衡常数的影响可用下式表示:
(4)
式中,T为热力学温度;为标准反应热效应。氨基甲酸铵分解反应是一个热效应很大的吸热反应,温度对平衡常数的影响比较灵敏。当温度在不大的范围内变化时,可视为常数,由(4)式积分得:
(C′为积分常数) (5)
若以对1/T作图,得一直线,其斜率为,由此可求出。并按下式计算T温度下反应的标准吉布斯自由能变化,
(6)
利用实验温度范围内反应的平均等压热效应和T温度下的标准吉布斯自由能变化,可近似计算出该温度下的熵变。
(7)
因此,通过测定一定温度范围内某温度的氨基甲酸铵的分解压,就可以利用上述公式分别求出:
四、实验内容与步骤
1.检漏 按图所示安装仪器。将烘干的小球和玻璃等压计相连,将活塞放在合适位置,开动真空泵,当测压仪读数约为53kPa,关闭三通活塞。检查系统是否漏气,待10min后,若测压仪读数没有变化,则表示系统不漏气,否则说明漏气,应仔细检查各接口处,直到不漏气为止。
2.装样品 确信系统不漏气后,使系统与大气相通,然后取下小球装入氨基甲酸铵,再用吸管吸取纯净的硅油或邻苯二甲酸二壬酯放入已干燥好的等压计中,使之形成液封,再按图示装好。
3.测量 调节恒温槽温度为(25.0±0.1)℃。开启真空泵,将系统中的空气排出,约15min后,关闭二通活塞,然后缓缓开启三通活塞,将空气慢慢分次放入系统,直至等压计两边液面处于水平时,立即关闭三通活塞,若5min内两液面保持不变,即可读取测压仪的读数。
4.重复测量 为了检查小球内的空气是否已完全排净,可重复步骤3操作,如果两次测定结果差值小于270Pa,方可进行下一步实验。
5.升温测量调节恒温槽温度为(27.0±0.1)℃,在升温过程中小心地调节三通活塞,缓缓放入空气,使等压计两边液面水平,保持5min不变,即可读取测压仪读数,然后用同样的方法继续测定30.0℃、32.0℃、35.0℃、37.0℃时的压力差。
6.复原 实验完毕,将空气放入系统中至测压仪读数为零,切断电源、水源。
五、思考题
1.测压仪读数是否是体系的压力?是否代表分解压?
2.为什么一定要排净小球中的空气?若体系有少量空气对实验有何影响。
3.如何判断氨基甲酸铵分解已达平衡?未平衡测数据将有何影响。
4.在实验装置中安装缓冲瓶的作用是什么?
5.玻璃等压计中的封闭液如何选择?
6.两者有何不同?
六、注意事项及其它说明
1.在实验开始前,务必掌握图中二个活塞(5和6)的正确操作。
2.必须充分排除净小球内的空气。
3.体系必须达平衡后,才能读取测压仪读数。
实验十一 原电池电动势的测定
一、实验目的
1.掌握可逆电池电动势的测量原理和电位差计的操作技术。
2.学会铜、锌等电极和盐桥的制备方法。
3.加深对原电池、电极电势等概念的理解
二、实验主要设备及使用要求
UJ25型电位差计;铜电极;锌电极;精密稳压电源;铂电极;电极管;饱和甘汞电极;标准电池
 UJ25型电位差计的使用:
UJ25型电位差计的使用:
(1)仪器控制面板示意图:见右图
(2)仪器操作步骤:
①组装好原电池后,按原电池的正、负极连接到仪器电池的“+”、“-”插孔。
图11-1 UJ25型电位差计控制面板
②把“选择电键”置于“校正”位置,用右下端的“零位调节”电位器调节使“调零显示屏”的示数为“0”。
③把“选择电键”转到“测量”位置,用平衡调节电位器的粗细调节电位器进行调节,再次使使“调零显示屏”的示数为“0”,调节过程中,先粗调,当“调零显示屏”的示数较小时,再进行细调,使“调零显示屏”示数再次为“0”,此时,“电动势显示屏”会显示一数据,该数据即为实验测定的原电池的电动势。
三、实验原理、方法和手段
凡是能使化学能转变为电能的装置都称之为电池(或原电池)。对定温定压下的可逆电池而言:
式中为法拉弟(Farady)常数,为电极反应式中电子的计量系数,为电池的电动势。
可逆电池应满足如下条件:
(1)电池反应可逆,亦即电池电极反应可逆
(2)电池中不允许存在任何不可逆的液接界。
(3)电池必须在可逆的情况下工作,即充放电过程必须在平衡态下进行,亦即允许通过电池的电流为无限小。因此,在制备可逆电池、测定可逆电池的电动势时应符合上述条件。
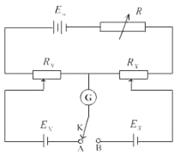 测量电池电动势最常用的方法是补偿法,在精确度不高的测量中,常用正负离子迁移数比较接近的盐类构成“盐桥”来消除液接电位,用电位差计测量电动势满足通过电池的电流为无限小的条件。其原理是在待测电池上并联一个大小相等、方向相反的外加电势,这样待测电池中就没有电流通过,外加电势差的大小就等于待测电池的电动势:
测量电池电动势最常用的方法是补偿法,在精确度不高的测量中,常用正负离子迁移数比较接近的盐类构成“盐桥”来消除液接电位,用电位差计测量电动势满足通过电池的电流为无限小的条件。其原理是在待测电池上并联一个大小相等、方向相反的外加电势,这样待测电池中就没有电流通过,外加电势差的大小就等于待测电池的电动势:
当电键K连接A时,有:
当电键连接B时,有:
因为,因此有:
图11-2对消法测定原电池电动势
可逆电池的电动势可看作正、负两个电极的电势之差,设正极电势为,负极电势为,则有:=-。
电极电势的绝对值无法测定,手册上所列的电极电势均为相对电极电势,即以标准氢电极作为标准(标准氢电极是氢气压力为101325Pa,溶液中各物质活度为1,其电极电势规定为零。将标准氢电极与待测电极组成一电池,所测电池电动势就是待测电极的电极电势。由于氢电极使用不便,常用另外一些易制备、电极电势稳定的电极作为参比电极。常用的参比电极有甘汞电极、银-氯化银电极等。这些电极与标准氢电极比较而得的电势已精确测出。
以铜锌电池为例,对铜电极可设计电池如下:
Zn(S)|ZnSO4(a1)‖CuSO4(a2)|Cu(S)
正极(铜电极)的反应为: Cu2++ 2e → Cu
负极(锌电极)的反应为: Zn → Zn2++ 2e
电池反应:Cu2++ Zn →Zn2++ Cu
相应的电极电势分别为:
= —
= —
所以,铜锌电池的电动势为:
=—=( —)-( —)
= [ —] —= —
纯固体的活度为1,则上式变为:
= —
式中,;:活度;:质量摩尔浓度;:硫酸铜溶液离子的平均活度系数。
四、实验内容与步骤
1.电极处理和制备
①锌电极的处理
将锌电极在稀硫酸溶液中浸泡5s,取出蒸馏水洗净后,再浸入汞或饱和硝酸亚汞溶液中约5s,表面上即生成一层光亮的锌汞齐,用蒸馏水冲洗后,滤纸搽干,插入装0.1000mol·kg-1ZnSO4溶液的电极管中待用。
②铜电极的制备
将铜电极在稀硝酸中浸泡5s,取出蒸馏水洗净,放入电镀装置中作为负极,以另一铜板作正极在镀铜液中电镀,控制电镀电流为20mA,电镀20min后得表面呈红色的Cu电极,蒸馏水洗净。滤纸搽干,插入装有0.1000mol·kg-1或0.0100 mol·kg-1CuSO4溶液的电极管中待用。
2.分别组成以下几个电池,用电位差计测定它们的电池电势:
Zn½ZnSO4(0.1000 mol/kg)½½KCl(饱和)½Hg2Cl2½Hg
Zn½ZnSO4(0.1000 mol/kg)½½CuSO4(0.1000 mol/kg)½Cu
Cu½CuSO4(0.0100 mol/kg)½½CuSO4(0.1000 mol/kg)½Cu
Hg½Hg2Cl2½KCl(饱和)½½CuSO4(0.1000 mol/kg)½Cu
五、思考题
讨论应用对消法测量电动势的优点。
六、注意事项及其它说明
1.仪器至少要预热半小时以后才能进行测定。
2.要迅速、准确进行电动势测定,避免因为极化作用造成较大的误差。
实验十二 希托夫法测定离子迁移数
一、实验目的
1.掌握希托夫法测定离子迁移数的原理及方法;
2.测定CuSO4水溶液中离子的迁移数;
3.学会使用电量计和分光光度计。
二、实验主要设备及使用要求
迁移管;铜电极;精密稳流电源;铜电量计;分析天平;7200分光光度计
三、实验原理、方法和手段
当电流通过电解质溶液时,溶液中的正负离子各自向阴、阳两极迁移,每种离子所带过去的电量与通过溶液的总电量之比,称为该离子在此溶液中的迁移数。
离子迁移数与浓度、温度、溶剂的性质有关,增加某种离子的浓度则该离子传递电量的百分数增加,离子迁移数也相应增加;温度改变,离子迁移数也会发生变化,但温度升高正负离子的迁移数差别较小;同一种离子在不同电解质中迁移数是不同的。
离子迁移数可以直接测定,方法有希托夫法、界面移动法和电动势法等。
希托夫法测定离子迁移数的示意图如图所示。将 图12-1希托夫法测定迁移数示意图
已知浓度的硫酸溶液装入迁移管中,若有Q库仑电量通过体系,在阴极和阳极上分别发生如下反应:
阳极: Cu-2e-→Cu2+
阴极: Cu2++2e-→ Cu
此时溶液中Cu2+离子向阴极方向迁移,SO2-4离子向阳极方向迁移。电极反应与离子迁移引起的总结果是阴
极区的CuSO4浓度减少,阳极区的CuSO4浓度增加。由于流过小室中每一截面的电量都相同,因此离开与进入假想中间区的Cu2+离子数相同,SO2-4离子数也相同,所以中间区的浓度在通电过程中保持不变。由此可得计算离子迁移数的公式如下:
式中,F为法拉第(Farady)常数,Q为总电量。
图12-1所示的三个区域是假想分割的,实际装置必须以某种方式给予满足。图12-2的实验装置提供了这一可能,它使电极远离中间区,中间区的连接处又很细,能有效地阻止扩散,保证了中间区浓度不变。
各极区CuSO4浓度可用分光光度计测定;阴极液通电前后CuSO4减少的量n可通过下式计算:
通过溶液的总电量可用铜电量计测定,根据法拉第定律及铜阴极
增加重量(m2-m1),得到求算总电量(库仑)公式如下:
图12-2 测定迁移数线路图
式中,F为法拉第(Farady)常数。
四、实验内容与步骤
1.准确配制为0.1mol·dm-3的CuSO4溶液250mL,,然后用该CuSO4溶液冲洗迁移管后,装满迁移管。
2.处理干净铜电极,准确称量铜电量计阴极铜片的重量。
3.按图接好线路,将稳流电源的“调压旋钮”旋至最小处。经教师检查后,接通开关K,打开电源开关,旋转“调压旋钮”使电流强度为10~15mA,通电约1.5h后,立即关紧两个活塞,然后关闭电源。
4.将铜电量计阴极铜片取下洗净、干燥,然后称重。中间液放入另一洁净干燥的烧杯中。
5.取10mL阴极液(或阳极液)放入三角瓶内,用7200分光光度计测定其吸光度。再取10mL中间液测定定,检查中间液浓度是否变化。
6.测定系列不同浓度CuSO4溶液的吸光度,以绘制标准曲线。
五、思考题
1.中间区浓度改变说明什么?如何防止?
2.为什么不用蒸馏水而用原始溶液润洗希托夫管?
3.影响本实验的因素有哪些?
六、注意事项及其它说明
电极使用前应打擦干净;通电过程中,迁移管应避免振动;中间管与阴极管、阳极管连接处不留气泡。
实验十三镍在硫酸溶液中的钝化行为
一、实验目的
1.学习和掌握金属钝化的原理和实验技术。
2.测量镍在硫酸溶液中的钝化行为。
3.学会处理电极表面,了解表面状态对钝化曲线的影响。
4.了解极化曲线的意义和应用。
二、实验主要设备及使用要求
LK98-II电化学工作站
三、实验原理、方法和手段
1.金属的阳极过程
当电极电势高于热力学平衡电势时,金属作为阳极将发生下面电化学溶解过程:
在金属的阳极溶解过程中,其电极电势必须高于其热力学电势,电极过程才能发生。这种电极电势偏离其热力学电势的现象称为极化。当金属上超电势不大时,阳极过程的速率随电极电势而逐渐增大,这是金属的正常溶解。但当电极电势正到某一数值时,其溶解速度达到最大,而后随着电极电势的变正,阳极溶解速度反而大幅度降低,这种现象称为金属的钝化现象。
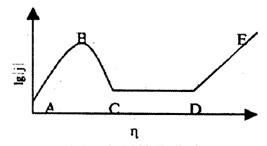 研究金属的阳极溶解及钝化过程通常采用控制电位法。对于大多数金属来说,其阳极极化曲线大都具有图1所示的形式。从恒电位法测定的极化曲线可以看出,它有一个“负坡度”区域的特点。具有这种特点的极化曲线是无法用控制电流的方法测定的。因为同一个电流I可能相应于几个不同的电极电势,因而在控制电流极化时,体系的电极电势可能发生振荡现象,即电极电势将处于一种不稳定状态。控制电位技术测得的阳极极化曲线(图13-1)通常分为四个区域:
研究金属的阳极溶解及钝化过程通常采用控制电位法。对于大多数金属来说,其阳极极化曲线大都具有图1所示的形式。从恒电位法测定的极化曲线可以看出,它有一个“负坡度”区域的特点。具有这种特点的极化曲线是无法用控制电流的方法测定的。因为同一个电流I可能相应于几个不同的电极电势,因而在控制电流极化时,体系的电极电势可能发生振荡现象,即电极电势将处于一种不稳定状态。控制电位技术测得的阳极极化曲线(图13-1)通常分为四个区域:
①活性溶解区(AB段):电极电位从初值 图13-1金属钝化曲线
开始逐渐往正变化,相应极化电流逐渐增加,此时金属进行正常的阳极溶解。
②过渡钝化区(BC段):随着电极电势增加到B点,极化电流达到最大值。若电极电位继续增加,金属开始发生钝化现象,即随着电势的变正,极化电流急剧下降到最小值。通常B点的电流Ip称为致钝电流,相应的电极电位称为致钝电位。在极化电流急剧下降到最小值的转折点(C点)电位称为Flade电位。
③稳定钝化区(CD段):在此区域内金属的溶解速度维持最小值,且随着电位的改变极化电流基本不变。此时的电流密度称为钝态金属的稳定溶解电流密度,这段电位区称钝化电位区。
④过钝化区(DE段):DE段为过钝化区,即钝化了的金属又开始溶解,电流密度随外加给定的电位的增加而迅速增大。
2.金属阳极溶解和钝化机理
金属的阳极极化过程是一复杂过程,包括活化溶解过程、钝化过程和过钝化过程等。它的机理还不是很清楚,以下描述可能对分析结果有所帮助。金属Me活化溶解是:
(1)
(2)
它的电流决定于中间物形成速度。将快速转变为。同时,阳极溶解可能同时发生:
(3)
产物按以下反应发生钝化过程:
(4)
(5)
钝化过程与反应(1)和(2)的活化溶解过程不同。它的双单电子串联过程,反应速度决定于表面的形成速度。随后快速转变为MeO,形成钝化层,阻滞继续溶解。溶液中H+离子会与钝化层物质产生化学反应,发生过钝化过程,即
(6)
溶液中阴离子(如离子)也能与钝层发生化学反应
(7)
产生可溶性,破坏钝化层,促使的活化溶解。
3.影响金属钝化过程的几个因素
金属钝化现象已进行了大量的研究工作。影响金属钝化过程及钝化性质的因素,可归纳为以下几点:
(1)溶液的组成。在中性溶液中,金属一般比较容易钝化,而在酸性或某些碱性的溶液中,则不易钝化;溶液中卤素离子(特别是Cl-)的存在,能明显地阻止金属的钝化;溶液中存在某些具有氧化性的阴离子(如CrO2-4)则可以促进金属的钝化。
(2)金属的化学组成和结构。各种纯金属的钝化能力不尽相同,例如铁、镍、铬三种金属的钝化能力为铬>镍>铁。因此,添加铬、镍可以提高钢铁的钝化能力及钝化的稳定性。
(3)外界因素(如温度、搅拌等)。一般来说,温度升高以及搅拌加剧,可以推迟或防止钝化过程的发生,这与离子扩散有关。
4.恒电势阳极极化曲线的测量原理和方法
控制电势法测量极化曲线时,一般采用恒电位仪,它能将研究电极的电势恒定地维持在所需值,然后测量对应于该电势下的电流。由于电极表面状态在未建立稳定状态之前,电流会随时间而改变,故一般测出的曲线为“暂态”极化曲线。在实际测量中,常采用的控制电势测量方法有下列两种。
①静态法:将电极电势较长时间地维持在某一恒定值,同时测量电流随时间的变化,直到电流值基本上达到某一稳定值。如此逐点地测量各个电极电势(例如每隔20,50或100mV)下的稳定电流值,以获得完整的极化曲线。
②动态法:控制电极电势以较慢的速度连续地改变(扫描),并测量对应电势下的瞬间电流值,并以瞬时电流与对应的电极电势作图,获得整个的极化曲线。所采用的扫描速度(即电势变化的速率)需要根据研究体系的性质选定。一般来说,电极表面建立稳态的速度愈慢,则扫描速率也应愈慢,这样才能使所测得的极化曲线与采用静态法接近。
上述两种方法都已获得了广泛的应用。从测定结果的比较可以看出,静态法测量结果虽较接近稳态值,但测量时间太长。本实验采用动态法。
四、实验内容与步骤
1.镍在硫酸溶液中钝化行为
本实验首先测量镍在硫酸溶液中阳极极化曲线,再观察氯离子对镍阳极钝化的影响。
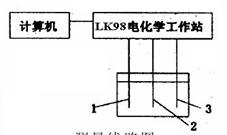 (1)了解仪器的线路及装置,并将线路接好,请教师检查(见图13-2)。
(1)了解仪器的线路及装置,并将线路接好,请教师检查(见图13-2)。
(2)将研究电极(电极)用金相砂纸磨至镜面光亮,然后在丙酮中清洗除油,电极面积为(用石蜡封多余面积),再用被测硫酸浸泡几分钟,除去氧化膜。
(3)洗净电极池,注入待测硫酸溶液,然后将研究电极、辅助电极、参比电极、盐桥和装入电极池内,通氮气 图13-2测量线路图
15分钟,除氧气。 1—辅助电极2参比电极3研究电极
(4)打开仪器和计算机的电源开关预热10分钟。
(5)在Windows98操作界面上运行“LK98-II”进入主控菜单。在“线性扫描伏安法”中设置参数:
初始电位:设为比先前所测得的开路电位负0.1伏
终止电位:设为1.7伏;
扫描速度:设为0.01伏/秒
等待时间:设为120秒
至此参数己设定完毕,点击“确定”键;然后点击工具栏中的运行键,此时仪器开始运行,屏幕上即是当时的工作状态和电流对电位的曲线。实验结束后,存盘,打印结果。
2.氯离子对镍阳极钝化的影响
更换溶液和重新处理研究电极。使Ni电极依次在0.5mol·L-1H2SO4+0.01mol·L-1KCl、0.5mol·L-1H2SO4+ 0.02mol·L-1KCl和0.5mol·L-1H2SO4+ 0.05mol·L-1KCl溶液中进行阳极极化。重复上述步骤进行测量。
实验完毕,关掉恒电位仪电源,再取出电极,清洗仪器。关闭计算机。
五、思考题
1.比较恒电位法和恒电流法所得到的极化曲线有何异同?说明原因。
2.测定阳极极化曲线为什么要用恒电位法。
3.做好本实验的关键是什么?
4.如要对某种系统进行阳极保护,首先必须明确哪些参数?
六、注意事项及其它说明
1.按照实验要求,严格进行电极处理。
2.实验前必须先测“参比”对“研究”电极的开路电位,合格时方能开始实验。
3.做完测试后,应在确认电化学工作站或电化学综合测试系统在非工作的状态下,关闭电源,取出电极。
实验十四 电导法测定乙酸的电离常数
一、实验目的
1. 学习电导率仪的使用方法。
2. 学会用电导法测定弱酸的电离常数。
3. 学会用工作曲线校正仪器的系统误差。
二、实验主要设备及使用要求
数字电导率仪
三、实验原理、方法和手段
因为温度一定时,弱电解质溶液的电离度满足关系:α ≈/,
而 ,
在25℃时乙酸的= 3.907×10-2S·m2/mol,
所以有乙酸的电离常数:
=
上式可变形为 ,
因为,所以上式变为
。
因此,将乙酸不同浓度时的与对应的作图,由直线的斜率即可求出电离常数。注意式中的
四、实验内容与步骤
1.用标准溶液配制浓度分别为0.0100,0.0010,0.0005 mol/l的KCl溶液各100ml。
2.用标准溶液配制浓度分别为0.0800,0.0500,0.0250,0.0100mol/l的乙酸溶液各100ml。
3.接通玻璃恒温槽及数字温度控制器的电源,开始搅拌,控温25℃。由于最后的测定结果将与25℃时的实验值对比,所以,当实际水温超过25℃时,不要打开恒温槽的加热器,只要打开搅拌器搅拌即可。在实际的水温下测定样品溶液的电导率,而且测定后须将此测得的电导率值校正为25℃时的数值后再用于后面的数据处理。校正比例:温度每升高1℃,测出的电导率值将平均比25℃时的数值增大1.9%。校正公式为:,式中t为测定电导率时的水浴温度(℃)。
4. 接通DDS-11C型数字电导率仪的电源,使其预热15分钟;并检查其电极连接是否正 常,实验中禁止触动电极与电导率仪之间的接口。
注意:(1)仪器上所读出的电导率的单位应为“μS/cm”或“mS/cm”(S读作“西门子”,1S = 1Ω-1),但仪器的盘面上所标示的单位只是“μS”和“mS”; (2)仪器面板上的数字表示该测量档位的理论最大量程。
5. 将电导率仪的温度补偿旋钮旋转到25℃,再将电导率仪的“电阻率/校正/电导率”旋钮旋到“校正”挡,旋转“常数”钮使仪器显示的数值(不计小数点)与电导池常数一致,仪器校正完毕;然后,再将仪器的“电阻率/校正/电导率”旋钮旋到“电导率”挡,将温补调到恒温水浴所控制的实际温度待用。
6. 将作电导池用的大试管及电极用蒸馏水洗净后装入约5ml蒸馏水置于恒温水浴中恒温5分钟后测其电导率,每隔20秒记录一个数据,连续记录三个值(同时要记录单位),取其平均值作为水的电导率值。读取电导率数值前需要测量的选择挡位,以获得最佳结果。一般要求所测的数值不超过该挡位所能测量的最大量程的1.5倍,同时又具有最多的有效数字位数,仪器不同测量挡位上方标明的数值表示该挡位的最大量程。
7. 测量所配样品溶液时,依浓度由小到大的顺序进行。取第一个样液分别润洗电极和大试管三次,而后向其中注入约5ml待测液,小心将电极插入试管的溶液里,使电极的铂片没于液面下,将试管连同电极一起放入恒温槽里恒温5分钟。
8. 每隔20秒读取电导率数值一次,共记录三个数据(同时要记录单位),取其平均值作为该样液的电导率值。
9. 小心地倒掉试管中的液体样品,并用蒸馏水洗净大试管和电极。并重复7、8的操作步骤直到测完其它样品溶液。
停止实验,先关闭各装置的电源开关,再拔下各仪器的插头,后取出、擦干数字式温控器、数显贝克曼温度计的探头;打扫小组实验台面的卫生,抄录一份原始实验数据记录,交教师签字后离开实验室。
五、思考题
1.实验中为什么要测定水的电导率?
2.对你实验结果的误差给予合理的解释。
六、注意事项及其它说明
1.实验中样品溶液的配制非常关键,一定要先洗净容量瓶,认真配准浓度,否则会严重影响实验的测定结果。
2.仪器一定要预热、校正,并进行温度补偿。
3.每个小组只有1-2支样品管作电导池使用,因此,在换溶液时,一定要事先洗净样品管、电极,并用待测液将样品管和电极淌洗三次。
4.操作过程中要十分小心,待用的电极一律要架在专用的电极支架上,以免损坏。
5.整个实验过程中恒温槽的温度都要保持恒定。
实验十五铁氰化钾在玻碳(金、铂)电极上的氧化还原
一、实验目的
1.学习固体电极表面的处理方法
2.学会电化学工作站的使用技术
3.了解可逆波的循环伏安图的特性,了解扫描速率和浓度对循环伏安图的影响。
4.掌握循环伏安法判断电极过程的可逆性,学会峰电流及峰电位的测量方法
二、实验主要设备及使用要求
DF—2000精密电化学分析工作站;工作电极:铂(金、玻碳)圆盘电极;辅助电极:铂丝电极;参比电极:饱和甘汞电极。
工作电极的预处理
没抛光过的新电极先用金相砂纸把电极表面磨平,然后用Al2O3(200-300)目抛光粉抛光电极表面,直至电极表面显像出镜面。最后分别在1∶1乙醇、1∶1 HNO3、蒸馏水中超声清洗(每次约5min ),取出用水洗净后置于0.5mol/LH2SO4溶液中,接通三电极系统,在-1.0-1.0V电位范围内,以1000mV/s的扫描速率进行循环扫描极化处理,至CV曲线稳定为止(约十周)。
三、实验原理、方法和手段
循环伏安法是在固定面积的工作电极和参比电极之间加上对称的三角波扫描电压,记录工作电极上得到的电流与施加电位的关系曲线,即循环伏安图。从伏安图的波形、氧化还原峰电流的数值及其比值、峰电位等可以判断电极反应机理。
将平面电极插入到如下电化学体系中,有如下反应:
式中O为氧化态物质,R为还原态物质。在电极上施加一随时间作线性变化的大幅三角波电位,如图15-1所示,这一电位叫做电位扫描。电位扫描以恒定的速度从到,然后再从返回到,在电位扫描作用下,通过电极的电流随电 图15-1电位扫描曲线
位而变,物质在电极上发生化学反应,得到电流-电位曲线,这个图叫做循环伏安曲线。电位负向扫描时,反应物O发生还原反应,,得
到的电流-电位曲线,叫做阴极极化曲线;电位正向扫描时,由于还原产物R来不及向溶液内部扩散,仍然保留在电极表面上,因而,产物R重新氧化为氧化态,,得到的电流-电位曲线叫阳极极化曲线,两根极化曲线,组成了循环伏安图,此方法称为循环伏安法。
循环伏安法通常采用三电极系统,即工作电极(被研究的物质起反应的电极,简写为WE);参比电极(监测工作电极电势的电极,简写为RE);辅助电极或者对电极(与工作电极组成让电流畅通的回路,简写为CE)。外加电压在工作电极与辅助电极之间,反应电流通过工作电极与辅助电极。
在本实验中,电极过程为可逆电极过程(电荷交换速度很快,反应由扩散控制的过程),对于本实验的氧化还原体系,当电压负向扫描时,在电极上还原,其反应为:
得到一个还原电流峰。当电压正向扫描时,在电极上氧化,其反应为:
得到一个氧化电流峰。所以,电压完成一次循环扫描后,将记录出如图( )所示的氧化还原曲线。
循环伏安法能迅速提供电活性物质电极反应的可逆性,化学反应历程,电活性物质的吸附以及电极有效表面积的计算等许多信息。
在循环伏安曲线中,阳极峰电流,阴极峰电流,阳极峰电位,阴极峰电位以及,是最为重要的参数。
对于一个可逆过程:
,一般情况下约为mV
,同时根据Randles-Sevcik公式,正向扫描的峰电流 为:
25°C时,
其中A为电极的有效面积(cm2),为反应物的扩散系数(cm2/s),为电极反应的电子转移数,为扫速(V/s),为反应物的浓度(mol/cm3),为峰电流(A)
从的表达式可以看出:与以及都成线性关系,这些特征可以作为判断一个电极过程是否可以的一个依据,对演剧电极过程具有重要意义,也可以根据此过程来计算电极的有效面积。
(本实验所用参数:电子转移数n=1,,的扩散系数)
四、实验内容与步骤
1.测试液配置
取一50ml容量瓶,用移液管依次加入1.0×10-2mol/LK3Fe(CN)6标准储备液2.5ml,
2.0mol/L的KNO3溶液5.0mL,用蒸馏水稀释至刻度,摇匀。
2.K3Fe(CN)6溶液的循环伏安曲线
取一定量的测试液于电解池(50ml烧杯)中(由电解池的大小来决定取试液的量,一般
取试液量为电解池容量的二分之一为宜),插入三电极,连接电极连线。在操作版面上选择循环伏安技术,设置扫描参数:起始电位600mV,终止电位-200mV,扫描速度为20mV/s,取样间隔2mV,量程100mA,通高纯氮气除氧10min(通氮气速度以明显看出溶液冒出连续气泡为宜),关闭氮气并让溶液静止30s,然后开始扫描,电脑同时记录I--E曲线,即为体系的循环伏安曲线(见图15-2)。
[K3Fe(CN)6]=5.0×10-4mol/L[KNO3]=0.2mol/L
扫描速度:100mV/s
电位范围:+600mV—-200mV
如果电极处理的好,该CV曲线具有可逆波的特性,其式量电位约为+0.23V(玻碳电极)。
3.不同扫描速度下K3Fe(CN)6溶液的循环伏安曲线
在上述体系改变扫描速度分别为40mV/s, 60mV/s, 80mV/s, 100mV/s, 其他条件不变,做不同扫描速度下溶液的循环伏安曲线,其峰电流应与扫描速度的二分之一次方成正比关系。
五、思考题
1.解释溶液的循环伏安图的形状。
2.如何利用循环伏安法判断电极过程的可逆性?
3.若实验中测得的条件电极电位和与文献值有差异,说明其原因。
六、注意事项及其它说明
1.实验结果的好坏与电极的处理有直接关系,一般来讲,电极处理的好,实验结果接近可逆反应的理论,否则,为准可逆反应。
2.每次扫描结束后,进行下次扫描前应将溶液搅动,防止电极表面的累积效应。
3.在扫描过程中,溶液应静止,避免扰动。
实验十六电导法测定乙酸乙酯皂化反应速率常数
一、实验目的
1. 掌握电导法测定反应速率常数的原理和方法,了解活化能的测定。
2. 用图解法验证二级反应的特点
3. 掌握电导率仪的使用方法
4. 理解物理性质在化学动力学实验中的应用。
二、实验主要设备及使用要求
数显电导率仪
三、实验原理、方法和手段
乙酸乙酯皂化反应是一个典型的二级反应
a b 0 0
反应速率方程为
式中:a ,b分别表示两反应物的初始浓度,x表示经过时间t后消耗的反应物浓度,k表示反应速率常数。为了数据处理方便,设计实验使两种反应物的起始浓度相同,即a=b,此时上式可以写成
积分得:
由(18.3)式可知,只要测得t时刻某一组分的浓度就可求得反应速率常数。测定该反应体系组分浓度的方法很多,例如,可用标准酸滴定测出不同时刻OH-离子的浓度。本实验使用电导率仪测量皂化反应进程中体系电导随时间的变化,从而达到跟踪反应物浓度随时间变化的目的。随着乙酸已酯皂化反应的进行,溶液中导电能力强的OH-离子逐渐被导电能力弱的CH3COO-离子取代,Na+离子浓度不发生变化,而CH3COOC2H5和C2H5OH不具有明显的导电性,所以溶液的电导逐渐减小,故可以通过反应体系电导的变化来度量反应的进程。
令G0、Gt和G∞分别表示反应起始时、反应时间t时和反应中了时反应体系的电导。显然是浓度为a的NaOH溶液的电导,是浓度为a的CH3COONa溶液的电导,Gt是浓度为(a-x)的NaOH溶液与浓度为x的CH3COONa溶液的电导之和。由此可得到下列关系式:
,整理可得
代入积分式得:
或
由以上两式可以看出,以对作图,或以对作图均可得一条直线,由直线斜率可求得速率常数,后者无需测得值。
已知, 故有或,所以,测出、、、 即可求出反应的速率常数。
若在不同得温度下测得反应速率常数,则可根据Arrhenius公式:
或
求得反应的活化能
四、实验内容与步骤
实验前打开恒温水浴,设定实验温度30℃,打开电导率仪,预热。
1.0.02mol.L-1CH3COOC2H5溶液配制(实验室已经配制)
先计算配制0.02M乙酸已酯100ml所需乙酸已酯的质量。在100ml容量瓶中加入20ml蒸馏水,用分析天平准确称重,然后用滴管滴入比计算量略重的乙酸已酯,加水至刻度,混合均匀后倒入干燥的锥形瓶中,再准确计算所称重量的乙酸已酯配成0.02mol.L-1溶液所需的体积,不足水量用刻度移液管补充加入。
2.测定
⑴仔细阅读实验室墙壁上的仪器使用说明,按电导率仪说明书校正仪器。
⑵取适量0.01mol.L-1NaOH溶液放入干净的大试管中,将电导电极插入塞好,置于恒温水浴槽中,恒温10分钟左右测定其电导率,直至读数稳定不变时为止,即为30℃时的
3.的测定(备选实验)
取适量0.01mol.L-1CH3COONa溶液放入干净的大试管中,将电导电极插入塞好,置于恒温水浴槽中,恒温10分钟左右测定其电导率,直至读数稳定不变时为止,即为30℃时的。
4.测定
取20ml 0.02mol.L-1CH3COOC2H5溶液和20ml 0.02 mol.L-1NaOH溶液分别注入两支干净的大试管中,置于恒温槽恒温约10分钟,然后将CH3COOC2H5倒入NaOH中,当约有一半CH3COOC2H5注入NaOH中后开始记时,作为反应起始时间。互相倾倒几次后使两种溶液充分混合,并使混合溶液注入其中的一支试管中,置于恒温槽中恒温并插入电导电极。从第6分钟开始读数,每隔2分钟记录一个,到第20分钟后每隔4分钟记录一个数据,一直记录到第50分钟为止。
5.注意事项
⑴空气中的CO2会溶入蒸馏水和配制的NaOH溶液而使溶液浓度发生改变。CH3COOC2H5溶液久置会缓慢水解。而水解产物之一,CH3COOH会部分消耗NaOH,所以,本实验所用蒸馏水应是新煮沸的,所配溶液应是新鲜配制的。
⑵电极不使用时应浸泡在蒸馏水中,用时用滤纸轻轻沾干水分(切勿擦拭电极中的铂片表面,以免影响电导池常数)。
五、思考题
1.反应进程中溶液的电导为什么发生变化?
2.为什么可以认为是0.01mol.L-1NaOH溶液的电导?
3.如果以对t作图求,还需知道,怎样测定简便?
4.本实验为何采用稀溶液,浓溶液可否?
5.本实验为何要在恒温条件下进行?
六、注意事项及其它说明
1.本实验需用电导水,并避免接触空气及灰尘杂质落入。
2.配好的NaOH溶液要防止空气中的CO2气体进入。
3.乙酸乙酯溶液和NaOH溶液浓度必须相同。
4.乙酸乙酯溶液需临时配制,配制时动作要迅速,以减少挥发损失。
实验十七 旋光法测定蔗糖水解反应速率常数
一、实验目的
1.学习旋光仪的使用方法
2.用旋光测定蔗糖在酸催化条件下的水解反应速率常数
二、实验主要设备及使用要求
旋光仪
三、实验原理、方法和手段
下列反应理论上是一个三级反应。
时间 C12H22O11(蔗糖) + H2O C6H6O6(果糖)+ C6H6O6(葡萄糖)总旋光度
t=0 C0 0 0
t=t C0-x x x
t= 0 C0 C0
但在低蔗糖浓度溶液中,即使蔗糖全部水解了,所消耗的水量也是十分有限的,因而H2O的浓度均近似为常数,而H+作为催化剂,其浓度是不变的,故上述反应变为准一级反应。其反应动力学方程应为:
即: 。
由于难以直接测量反应物的浓度,所以要考虑使用间接测量方法。因体系的旋光度与溶液中具有旋光性的物质的浓度成正比,所以有:
=
= [果糖是左旋性的,所以比例系数取负。]
=
所以有,
利用上述两式可得出C0与C0-x的比值,再将其代入上述动力学方程中,得:
即:
由此可见,实验中只要测出、、后即可作图求出,而反应的半衰期为t1/2= ln2/k1。
四、实验内容与步骤
1.阅读旋光仪的使用说明,掌握仪器的操作方法。
2.练习旋光仪的“零点”调节操作,练习旋光管的装液方法,理解旋光管放置于旋光仪中正确方法(试管外壁必须绝对擦干,旋光管的两端用镜头纸擦,中间用抹布擦),了解旋光仪的读数方法。
3.使用所给的盐酸标准溶液在有刻度的试管中配制1.8mol/L的盐酸溶液50ml并加盖。粗略称量10克蔗糖在有刻度的试管中配制50ml溶液(称量蔗糖时注意不要泼撒在桌面上)。
4.将装有蔗糖溶液的试管、盐酸溶液的试管同时放入25℃的水浴中恒温10分钟。
5.每一个实验小组派一名同学领取旋光试管。
6.室温下、的测定。取出恒温后的两支试管,擦干管外壁的水珠后,将盐酸注入到蔗糖溶液中,当约有一半盐酸被注入到蔗糖溶液中时开始记时(这个时间近似当做是反应开始的时间),迅速将溶液充分混匀。随后立即用少量混匀的样品溶液淌洗旋光试管,装样、擦净试管外壁并放入旋光仪中。(同时由一个同学进行“7”的操作。)从开始记时算起,反应到大约第3分钟时读取第一个数据,然后分别在第5、7、9、12、14、16、18和20分钟时各记录一个值,此后每隔5分钟记录一次,一直记录到旋光度出现第一个负值为止。注意:在测量记录的同时,要每隔10分钟测量记录一次旋光仪中放置旋光试管的光路的温度,最后将所测得的各温度平均值作为蔗糖水解温度的近似值。
7.室温下的测定。将测定时剩余的溶液倒入洁净的大试管里,并置于55℃恒温水浴中30分钟,然后冷却到室温再装管测定三次旋光度取平均值(即)。
8.实验完毕后,关闭电源。小心地清洗仪器,尤其是旋光管要清洗干净(至无酸性为止),且注意千万不要将旋光管两头的玻片弄丢了。领取旋光试管的同学退回试管。打扫小组实验台面的卫生,抄录一份原始实验数据记录,交教师签字后离开实验室。
五、思考题
1.实验中先用蒸馏水校正旋光仪的零点,在蔗糖转化反应过程中所测的αt是否需要零点校正?为什么?
2.改变蔗糖溶液的初始浓度和催化剂盐酸的浓度对测量出的速率常数k和半衰期t1/2值有无影响?为什么?
3.如果实验所用蔗糖不纯,对实验有什么影响?
六、注意事项及其它说明
1.实验中先用蒸馏水校正旋光仪的零点,在蔗糖转化反应过程中所测的αt是否需要零点校正?为什么?
2.改变蔗糖溶液的初始浓度和催化剂盐酸的浓度对测量出的速率常数k和半衰期t1/2值有无影响?为什么?
3.如果实验所用蔗糖不纯,对实验有什么影响?
实验十八 丙酮碘化反应速率方程的测定
一、实验目的
1.测定用酸作催化剂时丙酮碘化反应的反应级数、速率常数及活化能。
2.初步认识复杂反应机理,了解复杂反应的表观速率常数的求算方法。
3.进一步掌握分光光度计的使用方法。
二、实验主要设备及使用要求
721型分光光度计
三、实验原理、方法和手段
酸催化的丙酮碘化反应是一个复杂反应,在反应的初始阶段,反应为:
+++
(A) (E)
该反应由催化,而反应本身又能生成,所以这是一个自催化反应,其速率方程为:
=== (1)
式中,为碘化丙酮的浓度;为氢离子的浓度;为丙酮的浓度;表示丙酮碘化反应总的速率常数,、、为反应级数。
可见,只要能测定反应过程中某一物质各时刻的浓度,就可以求出。由于碘在可见光区有一个比较宽的吸收带,所以可利用分光光度计来测定丙酮碘化反应过程中碘的浓度,从而求出反应的速率常数。
按照朗伯-比耳(Lambert-Beer)定律,透光率与碘的浓度之间的关系可表示为: =- (2)
式中,为透光率,为比色槽的光径长度,为摩尔吸收系数。
(2)式对反应时间求导,可得:
==-() (3)
式中可通过测定一已知浓度的碘溶液的透光率测出,由(3)式得:
==
作~关系图,得到一条直线,由直线斜率结合测定出来的值,可以求得反应体系的反应速率。
要测定一种物质的反应级数,至少需要进行两次实验,两次实验中,其他物质的初始浓度保持相同只改变该物质初始浓度进行动力学实验,将会得到该物质在反应条件下的反应级数,这种测定方法称为孤立法。例如级数a为丙酮的级数,若要求出a的具体数值,则要进行两次实验,若用“I”、“II”分别表示这两次实验:
(I),,;
(II),,;
由上述实验方法和计算分别测定得到的反应速率分别为,,则有:
= (4)
= (5)
对(4)、(5)两式取对数,得:
=+++ (6)
=++++ (7)
(7)-(6)得:
-=
=
=(/)
同理可以求出、,各反应体系的速率常数可以通过前面测定出来的数据代入(1)式中进行计算,总速率常数为这些测定体系速率常数的平均值。
由两个或两个以上温度的速率常数,就可以根据阿累尼乌斯(Arrhenius)关系式估算反应的活化能。:
Ea = 2.303R (T1T2)/(T1—T2) lg(k2/k1)
或Ea = R (T1T2)/(T1—T2) ln(k2/k1) (8)
四、实验内容与步骤
1.接通721型分光光度计的电源,选择入射光波长为565nm,灵敏度为“2”或“3”,打比色皿暗盒盖,调节“0”电位器使电表指针为“0”,然后关上暗盒盖,比色皿座处于蒸馏水校正位置,调节“100%”电位器,使电表指针达到满刻度附近,仪器预热20min。
2.将I2标准溶液稀释成浓度为0.0010M的溶液,测定它的透光率,测三次,取平均值。
3.按照下表中体积,分别移取各标准溶液于50ml容量瓶中,当最后一种物质加入体系的体积为应加入体积的时,开始开动停表记时,迅速用蒸馏水定容至刻度,混合均匀后用721型分光光度计测定不同时刻各动力学体系的透光率,体系1、2每1min/次,3、4每0.5min/次,直至体系透光率示数为95%时结束实验
表18-1 各动力学体系反应物配比
体系
| I2标准溶液
/ml
| CH3COCH3标准溶液/ml
| H+标准溶液
/ml
|
1
2
3
4
| 10
10
10
5
| 10
5
10
10
| 5
5
10
5
|
五、思考题
如何测定得到准确的实验结果?
六、注意事项及其它说明
1. 分光光度计要预热15min后才能进行测定。
2. 仪器处于工作状态时,不能打开样品箱盖。
3. 注意用蒸馏水为参比,调节仪器的“0”和“100%”透光率。
4. 反应混合顺序为:先加盐酸、丙酮,在容量瓶中,预留出一定体积的空间后,先加一定量的蒸馏水,平摇混匀后,再加入碘。
5. 配置样品要准确。
实验十九 H2O2催化分解反应速率常数的测定
一、实验目的
1.测定H2O2催化分解反应速率常数;
2.学会恒温槽的调节方法。
二、实验主要设备及使用要求
氧气的测量装置
三、实验原理、方法和手段
H2O2的化学性质是在常温的条件下缓慢分解,在有催化剂的条件下,分解速率明显加快,其反应的方程式如下:
H2O2= H2O+1/2O2
在有催化剂的条件下,其反应机理为:
H2O2+KI→KIO+ H2O (1)
KIO→KI+O2 (2)
 其中(1)的反应速度比(2)的反应速度慢,所以H2O2催化分解反应的速度主要由(1)决定,如果假设该反应为一级反应,其反应速度式如下:
其中(1)的反应速度比(2)的反应速度慢,所以H2O2催化分解反应的速度主要由(1)决定,如果假设该反应为一级反应,其反应速度式如下:
-dCH2O2/dt=KCkI.CH2O2
在反应的过程中,由于KI不断再生,故其浓度不变,上式可以简化为:
-dCH2O2/dt=K1CH2O2
积分后可以得到IgCt/C0=-K1/2.303.t
C0--- H2O2的初始浓度
Ct----反应到t时刻的H2O2浓度 图19-1 H2O2分解O2体积测量系统示意图
K1----KI作用下,H2O2催化分解反应速率常数
关于t时刻的H2O2浓度的求法有许多种,本实验采用的是通过测量反应所生成的氧的体积量来表示.因为在分解的过程中,在一定时间内,所产生的氧的体积与已分解的H2O2浓度成正比,其比例常数是一定值即
Ct=K(V∞-Vt)
C0=K V∞
V∞--H2O2全部分解所产生的氧气的体积
Vt----反应到t时刻时,所产生的氧气的体积
式中K为比例常数,将此式代入速率方程式中,可得到
IgCt/C0=Ig(V∞-Vt)/ V∞=-K1/2.303.t改写为
Ig(V∞-Vt)=- K1/2.303.t+IgV∞
如果以t为横轴,以Ig(V∞-Vt)为纵轴,可以得到一直线,即可验证H2O2催化分解反应为一级反应,由直线的斜率即可求出速率常数K1值.
而V∞可通过测定H2O2的初始浓度,利用计算的方法得到.
公式如下:
V∞=MH2O2VH2O2RT/2.P
P---氧的分压,由大气压减去实验温度下,水的饱和蒸汽压,
MH2O2----H2O2的初始浓度
VH2O2------实验中所取用的H2O2的体积
R----气体常数
T-----实验温度K
四、实验内容与步骤
1.调恒温槽为25C左右;
2.装置反应液
首先在小烧瓶中加入20mL的蒸馏水,再加入10mL实验用的H2O2溶液,再在试管中加入10mLKI,将小烧瓶与试管均放入恒温槽中预热5分钟;
3.调整测量系统
首先将氧气测量装置中的三通阀门,置于三通的位置,打开弹簧夹,提高水位瓶,使量气管的水位上升到0刻度,再关闭弹簧夹,将水位瓶放置桌面上;
4.开始催化分解反应
由一名同学将试管里的KI迅速到入小烧瓶中,塞紧胶塞,放于恒温槽中,匀速摇动.另一名同学同时将三通至于与外界隔断的位置,并同时启动秒表记时.并将平衡管的水位下降5厘米左右.
5.测量氧气的体积
当平衡管的水位与量气管的水位一平时,记下量气管的刻度.再打开弹簧夹,将平衡管的水位下降5厘米左右.再继续测量.共测量5次.
6.MnO2催化反应
KI催化分解以做完之后,要求按上述方法再做一遍MnO2催化反应,所取的H2O2的浓度,体积和水的数量同前,但加入的催化剂为MnO2,数量为一满耳勺,在H2O2预热后,直接加入。
7.测量H2O2的初始浓度
取5mL实验用的H2O2于锥形瓶中,再加入10mL3 MH2SO4用KMnO4溶液进行滴定,当溶液出现淡红色时为反应终点,记下消耗的KMnO4的体积.
五、思考题
在反应的过程中,摇动的快慢对速率常数的测定有没有影响?为什么?
六、注意事项及其它说明
1.为保证测试温度,本仪器在使用前必须开机预热半小时
2.内筒中加一定体积的水后若有气泡逸出,说明氧弹漏气,设法排除。
3.搅拌时不得有摩擦声。
4.燃烧样品萘时,内筒水要更换且需重新调温
5.每次实验结束,要小心地拿出测温探头,再打开量热计的上盖,以免损坏测温探头。把所用仪器如氧弹、坩锅、内桶等清洗干净并放回原处。
6.顺时针旋转氧气钢瓶阀杆,是关好钢瓶。反时针旋转减压阀杆,是关好减压阀。要特别注意安全。开时先开氧气钢瓶,再开减压阀;关时先关钢瓶,再关减压阀。实验过程中,不用频繁开关钢瓶和减压阀。实验结束时,才要关好钢瓶和减压阀。(为什么?)
7.实验过程中不要做与本实验无关的事,不要动与本实验无关的仪器。
实验二十 最大泡压法测定溶液的表面张力
一、实验目的
1.了解表面自由能、表面张力的意义及表面张力与吸附的关系。
2.掌握最大气泡法测定表面张力的原理和技术。
3.通过测定不同浓度正丁醇水溶液的表面张力,计算吉布斯表面吸附量和乙醇分子的横载面积。
4.学会以镜面法作切线,并利用吉布斯吸附公式计算不同浓度下正丁醇溶液的表面吸附量。
5.求正丁醇分子截面积和饱和吸附分子层厚度。
二、实验主要设备及使用要求
DP-A精密数字压力计
三、实验原理、方法和手段
液相与气相之间的界面层可看作是介乎液体与气体性质的第三相。界面层分子受液体内部分子的吸引力远大于外部蒸气分子对它的吸引力,致使表面层分子受到向内的拉力而导致表面积趋于最小(球形),以达到受力平衡。揭示表面层这一特征的方法很多,最常用的为表面张力(surface tension,用或表示),其定义是:抗拒表面积扩展单位长度的力,法定单位为N×m-1,也常用dyn×cm-1,或可定义为单位表面吉布斯自由能(surface Gibbs free energy,用表示),单位为kJ•m-2。表面张力和表面吉布斯自由能分别是用力学方法和热力学方法研究表面现象时采用的物理量,液体的表面张力和表面吉布斯自由能在数值上是相等的,但是具有不同的物理意义。
液体的表面张力与温度、纯度等因素有关。温度愈高,表面张力愈小;纯度发生变化时,表面张力也相应发生变化,其变化的大小决定于溶质的本性和加入量的多少。
根据能量最低原理,若溶液质能降低溶剂的表面张力,则表面层溶质的浓度应比溶液内部的浓度大;如果所加溶质能使溶剂的表面张力增加,那么,表面层溶液质的浓度应比内部低,这种现象为溶液的表面吸附,用吉布斯(Gibbs)公式表示:
式中,为表面吸附量(mol×m-2);为表面张力(J×m-2);为绝对温度(K);为溶液浓度(mol×L-1); 表示在一定温度下表面张力随浓度的变化率。
< 0,>0,溶质能增加溶剂的表面张力,溶液表面层的浓度大于内部的浓度,称为正吸附作用。
>0,<0 ,溶质能增加溶剂的表面张力,溶液表面层的浓度小于内部的浓度,称为负吸附作用。
可见,通过测定溶液的浓度随表面张力的变化关系可以求得不同浓度下溶液的表面吸附量。吸附量与浓度之间的关系可以用Langmuir等温吸附方程式表示:
式中,表示吸附量,通常指单位质量吸附剂上吸附溶质的摩尔数;表示饱和吸附量;表示吸附平衡时溶液的浓度;为常数。将上式整理可得如下形式:
作 ~ 图,得一直线,由此直线的斜率和截距可求常数和。
如果以N代表1m2表面层的分子数,则:
式中,为Avogadro常数,则每个分子的截面积A为:

测定表面张力的方法很多,有最
大泡压法、拉环法、张力计法等,在
本实验中,采用最大泡压法来测定液
体的表面张力。装置图如图20-1。
待测液体置于支管试管中,使毛
细管端面与液面相切,液面随着毛细 图20-1 实验装置图
管上升至一定高度。打开滴液漏斗缓慢抽气。此时,由于毛细管液面所受压力大于支管试管液压力(即有一压力差产生——附加压力ΔP=P大气-P系统),当在毛细管端面上产生的作用力稍大于毛细管口液体的表面张力时,毛细管液面不断下降,气泡就从毛细管口脱出。
毛细管内液面上受到一个比支管试管中液面上大的压力,当此压力差——附加压力(P大气-P系统)在毛细管端面上产生的作用力稍大于毛细管口液体的表面张力时,气泡就从毛细管口脱出,此附加压力与表面张力成正比,与气泡的曲率半径成反比,其关系式为:
。
如果毛细管半径很小,则形成的气泡基本上是球形的。当气泡开始形成时,表面几乎是平的,这时曲率半径最大;随着气泡的形成,曲率半径逐渐变小,直到形成半球形,这时曲率半径R和毛细管半径r相等,曲率半径达最小值,根据上式这时附加压力达最大值。气泡进一步长大,R变大,附加压力则变小,直到气泡逸出,根据上式,R=r时的最大附加压力为:
在实验中,如果使用同一支毛细管和压力计,则可以用已知表面张力的液体作为标准,分别测定它们的最大附加压力后,通过对比计算得到其它未知液体的表面张力:
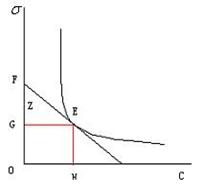 本实验以蒸馏水为标准物质,先测定水的最大附加压力值,查表得到实验温度下的,则
本实验以蒸馏水为标准物质,先测定水的最大附加压力值,查表得到实验温度下的,则
作图(图20-2),求出浓度下的值,则
图20-2 待测样品的表面张力-浓度图
结合前面的计算公式和处理方法,则可计算出G∞和A。
四、实验内容与步骤
1.正丁醇的密度测量:取一干净的称量瓶,准确称量空重W1后;装满蒸馏水在30℃恒温槽中恒温15min,迅速擦干外表面,并准确称量其质量W2;干燥称量瓶后,装满正丁醇在30℃恒温槽中恒温15min,迅速擦干外表面,准确称量其质量W3。结合30℃水的密度,计算出正丁醇在30℃的密度。
2.分别移取0.20ml,0.40ml,0.80ml,1.2m,l.6ml的正丁醇到容量瓶中,配制成100ml待侧液,各待侧液浓度结合正丁醇的密度和体积计算出来。
3.仔细清洗表面张力仪,调节恒温槽的温度为30℃,测定蒸馏水的最大附加压力△Pmax,测定3次,取平均值。
4.依次测定各浓度正丁醇水溶液的最大附加压力△Pmax值
五、思考题
1.在测量中,如果抽气速率过快,对测量结果有何影响?
2.如果将毛细管末端插入到溶液内部进行测量行吗?为什么?
3.本实验中为什么要读取最大压力差?
4.表面张力仪的清洁与否与温度的不恒定对测量数据有何影响?
六、注意事项及其它说明
1.仪器系统不能漏气。
2.测定过程中严格控温,配样品时,应在30℃下配制,因此,用移液管移取适量正丁醇到100ml容量瓶后,加入适量蒸馏水使液面刻度距离容量瓶刻度3厘米,放入恒温槽中恒温10分钟后,用恒温好的蒸馏水定容至刻度线,并混匀。
3.测定时,待测液浓度要按由稀到浓的顺序测定。
4.测定用的表面张力计一定要清洁,尤其是毛细管部分一定要干净,应保持垂直,其管口刚好与液面相切,否则气泡不能连续逸出,使压力计的读数不稳定,且影响溶液的表面张力。
5.读取压力计的压差时,应取气泡单个逸出时的最大压力差。
实验二十一 固体在溶液中的吸附
一、实验目的
1.测定活性炭在醋酸水溶液中对醋酸的吸附作用,并由此计算活性炭的比表面;
2.验证弗罗因德利希(Freundlich)经验公式和兰格缪尔(Langmuir)吸附公式;
3.了解固-液界面的分子吸附。
二、实验主要设备及使用要求
碱式滴定管;恒温振荡机
三、实验原理、方法和手段
活性炭是用途广泛的吸附剂,除了用于吸附气体以外,也用于溶液中的吸附,通常以克吸附剂吸附溶质的量来表示吸附量,在恒定温度的条件下,吸附量与溶液中吸附质的平 衡浓度有关,弗罗因得利希从吸附量和平衡浓度的关系曲线得出经验方程
X/m=KC1/n (1)
X------吸附溶质的量 mol
m-------吸附剂的量 g
C-------平衡浓度mol/L
K. n---常数,由温度.溶剂.吸附质及吸附剂的性质决定
将(1)式取对数可得
LgX/m=1/nLgC+LgK
以LgX/m为纵轴,以LgC为横轴,可得一直线,从直线的斜率和截距可求得K. n
四、实验内容与步骤
1.准备6个干的编好号的带塞的锥形瓶,按表格规定的浓度配置醋酸溶液,注意时塞好瓶塞,以防醋酸的挥发.
2.将烘干的活性炭装在称量瓶中,用差减法称取活性炭各左右1克左右,分别放于各号溶液中,塞好瓶塞,在振荡机上振荡1小时.
3.取2毫升实验用的醋酸,,加入酚酞指示剂,用0.1MNaOH溶液滴定,测量醋酸的初始浓度.
4.按记录表规定的体积进行取样分析,加入到锥形瓶中,再加入2滴酚酞指示剂,用0.1MNaOH溶液滴定到溶液出现淡红色,记下NaOH消耗的体积.
5.将用过的活性炭回收到回收瓶内.
五、思考题
1.吸附作用与哪些因素有关?固体吸附剂吸附气体与从溶液中吸附溶质有何不同?
2.试比较弗罗因德利希吸附等温式与兰缪尔吸附等温式的优缺点?
3.如何加快吸附平衡的到达?如何判定平衡已经到达?
4.讨论本实验中引入误差的主要因素?
六、注意事项及其它说明
1.温度及气压不同,得出的吸附常数不同
2.使用的仪器干燥无水;注意密闭,防止与空气接触影响活性炭对醋酸的吸附。
3.滴定时注意观察终点的到达。
4.在浓的HAc溶液中,应该在操作过程中防止HAc的挥发,以免引起较大的误差。
5.本实验溶液配制用不含CO2的蒸馏水进行。
实验二十二 表面活性剂CMC值的测定
一、实验目的
1.了解表面活性剂临界胶束浓度的测定原理。
2.掌握临界胶束浓度的测定方法和表面张力仪的使用方法。
3.掌握用电导法测定临界胶束浓度的方法。
二、实验主要设备及使用要求
HSS-1B数字式超级恒温浴槽、DDS-11A型电导率仪

图22-1 电导率仪
三、实验原理、方法和手段
表面活性剂分子:具有亲水性的极性基团和具有憎水性的非极性基团所组成的有机化合物。当它们以低浓度存在于某一体系中时,可被吸附在该体系的表面上,采取极性基团向着水,非极性基团脱离水的表面定向,从而使表面自由能明显降低。
在表面活性剂溶液中,当溶液浓度增大到一定值时,表面活性剂离子或分子不但在表面聚集而形成单分子层,而且早溶液本体内部也三三两两的以憎水基相互靠拢,聚在一起形成胶束。形成胶束的最低浓度称为(Critical Micelle Concentration CMC)。
表面活性剂随浓度变化的物理化学性质都可以用于测定CMC。
在CMC点,因溶液结构改变,其物理性质,化学性质明显转折。(表面张力,电导率,渗透压,浊度,光学性质).
作为表面活性剂表面活度的一种量度。临界胶束浓度小,但激发形成胶束所需浓度小,达到表面饱和吸附的浓度小,改变表面性质所需浓度小。临界胶束浓度是溶液性质显著变化的“分水岭”。
常用的方法:表面张力法、电导法、染料法等。
本实验采用电导法测定表面活性剂的电导率来确定CMC值。它是利用离子型表面活性剂水溶液的电导率随浓度的变化关系,作Λm-C1/2曲线,由曲线的转折点求出CMC值。
Λm=κ/C Λm(S·m2/mol),C(mol/L)
若温度恒定,在极稀的浓度范围内,强电解质溶液的摩尔电导率Λm与其溶液浓度的c1/2成线形关系。
对于胶体电解质,在稀溶液时的电导率,摩尔电导率的变化规律与强电解质一样,但是随着溶液中胶团的生成,电导率和摩尔电导率发生明显变化,这就是确定CMC的依据。
四、实验步骤
1.准备工作
1)打开超级恒温槽电源,将温度调到30℃,并将装有0.02M十二烷基磺酸钠的溶液的试剂瓶放入水浴中,以使试样完全溶解,当溶液澄清后取出。
2)将电导电极用蒸馏水洗净,并擦干备用。
3)打开电导率仪开关,将高低周换向开关调向“低周LOWF”,预热15min。
4)在11支比色管中分别移取1.25、2.5、5.0、7.5、10.0、12.5、15.0、17.5、20.0、22.5、25.0mL0.02mol/L十二烷基磺酸钠溶液,分别配制0.001、0.002、0.004、0.006、0.008、0.01、0.012、0.014、0.016、0.018、0.02mol/L的待测溶液,然后分成两组用皮筋扎起后放入恒温槽中恒温15min.
5)“校正-测量”开关调向“校正CHECK”,“调节ADJ”校正旋钮使仪器指针指向满刻度。

图22-2 电导率仪板面图
K1:电源开关 K2:高周、低周开关 K3:校正、测量开关
K4:校正调节 K5:量程选择开关 K6:电容补偿调节
K7:电极常数调节 C1:电极插口 C2:10毫伏输出插口
2.测量工作
1)把“校正-测量”开关调向“测量”档。将电极放入比色管中,用电导率仪测定样品的电导率,调节K5“量程选择开关”,选择合适量程,稳定,按相应量程“红”,“黑”读数,记录数据。
2)每次测完要把“校正-测量”开关调到“校正ADJ”后再从试样中取出电极;
3)在测定每个样品之前电导电极必须清洗并擦干。
4)按照浓度由低到高的顺序依次测定.
5)测完后关闭电源,并弃去被测液。
6)以Λm—C1/2作图得到CMC值。
五、思考题
1.电导法测定表面活性剂电导率确定CMC值的优点和适用范围。
2.该实验有多处需要加热,且时间较长,如何统筹安排,节约时间。
3.引起实验结果误差有哪些主要原因。
六、注意事项及其它说明
1.离子型药品要分析纯,易溶。称样前须烘干(不烤焦)不含水等其它杂质
2.摩尔系列溶液定容配制要准确。
3.注意预温、恒温测定。
4.测准电导电极的仪器常数,校正电导率仪并正确进行测溶液电导的操作。
5.清洗电导电极时,两个铂片不能有机械摩擦,可用电导水淋洗,后将其竖直,用滤纸轻吸,将水吸净,并且不能使滤纸沾洗内部铂片。
6.注意电导率仪应有低到高的浓度顺序测量样品的电导率,以减小实验误差。
7.电极在冲洗后必须擦干,以保证溶液浓度的准确,电极在使用过程中其极片必须完全浸入到所测的溶液中。
8.一次测量结束后要将“校正-测量”开关调至“校正”处。
9.溶液未变澄清之前,不可将溶液分份,因浓度不均匀。
10.电导法测定表面活性剂电导率,过量无机盐使其灵敏度下降(配制溶液时使用电导水)。
实验二十三 B-Z振荡反应
一、实验目的
1.了解、熟悉化学振荡反应的机理
2.通过测定电位-时间曲线求得化学振荡反应的表观活化能
3.掌握计算机在化学实验中的应用
二、实验主要设备及使用要求
电化学分析仪
三、实验原理、方法和手段
人们通常所研究的化学反应,其反应物和产物的浓度呈单调变化,最终达到不随时间变化的平衡状态。而某些化学反应体系中,会出现非平衡非线性现象,即有些组分的浓度会呈现周期性变化,该现象称为化学振荡。为了纪念最先发现、研究这类反应的两位科学家(BelouS0v和Zhabotinskii),人们将可呈现化学振荡现象的含溴酸盐的反应系统笼统地称为BZ振荡反应(Bz Oscillating Reaction)。
大量的实验研究表明,化学振荡现象的发生必须满足3个条件:(1)必须是远离平衡的敞开体系;(2)反应历程中应含有自催化步骤;(3)体系必须具有双稳态性(bistability),即可在两个稳态间来回振荡。
有关BZ振荡反应的机理,目前为人们所普遍接受的是FKN机理,即由Field、Kоrоs和Noyes三位学者提出的机理。对于下列著名的化学振荡反应
(A)
FKN机理认为,在硫酸介质中以铈离子作催化剂的条件下,丙二酸被漠酸盐氧化的过程至少涉及9个反应。
1.当上述反应中[Br-]较大时,BrO3-是通过下面系列反应被还原为Br2的:
 (1)
(1)
 (2)
(2)
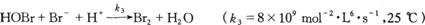 (3)
(3)
其中反应(10.A)是控制步骤。上述反应产生的Br2使丙二酸溴化
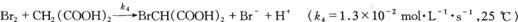 (4)
(4)
因此,导致丙二酸溴化的总反应(10.1)为上述四个反应之和而形成一条反应链,
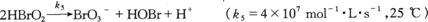 (α)
(α)
2.当[Br-]较小时,溶液中的下列反应导致了铈离子的氧化
(5)
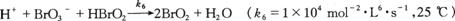 (6)
(6)
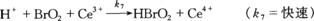 (7)
(7)
上面三个反应的总和组成了下列反应链,
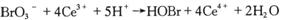 (β)
(β)
该反应链是振荡反应发生所必需的自催化反应,其中反应式(Ⅱ一15—6)是速度控制步骤。
最后,Br-可通过下列两步反应而得到再生,
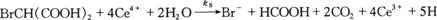
 (8)
(8)
上述两式偶合给出的净反应为:
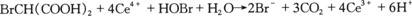 (γ)
(γ)
如将反应式(α)、(β)和(γ),)相加就组成了反应系统中的一个振荡周期,即得到总反应式(A)。必须指出,在总反应中铈离子和溴离子已对消,起到了真正的催化作用。
综上所述,BZ振荡反应体系中存在着两个受溴离子浓度控制的过程(α)和(β),即[Br-]起着转向开关的作用,当[Br-]>临界浓度[Br-]临界时发生(α)过程;而当[Br-]< [br-]临界时发生(β)过程。该反应溴离子的临界浓度为:
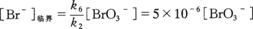
若已知实验的初始[Br03-],由上式可估算[Br-]临界。
测定、研究BZ化学振荡反应可采用离子选择性电极法、分光光度法和电化学等方法。本实验采用电化学方法,即在不同的温度下通过测定因[Ce4+]和[Ce3+]之比产生的电势随时间变化曲线,分别从曲线中(如图10.1)得到诱导时间(tu)和振荡周期(tZ),并根据阿仑尼乌斯(Arrhenius)方程,
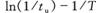 ,式中E为表观活化能;R是摩尔气体常数(8。314 J·mol-1·K-1;T是热力学温度;A是经验常数。分别作
,式中E为表观活化能;R是摩尔气体常数(8。314 J·mol-1·K-1;T是热力学温度;A是经验常数。分别作
和In(1/tZ)一1/T图,最后从图中的曲线斜率分别求得表观活化能(EU和EZ)。
四、实验内容与步骤
1.配制溶液
分别用蒸馏水配制0.005 mol·L-1硫酸铈铵(必须在0.2 mol·L-1H2S04硫酸介质中配制)、0.4 mol·L-1丙二酸、0.2 mol·L-1溴酸钾、3 mol·L-1硫酸各100 mL。
 2.准备工作
2.准备工作
(1)测量线路如图23-1所示。打开仪器电源预热10min;同时开启恒温槽电源(包括加热器的电源),并调节温度为30℃(或比当时的室温高3-5℃)。
(2)将配好的硫酸铈铵、丙二酸和硫酸溶液各10 mL放入已洗干净的电解池中,同时也将10 mL溴酸钾溶液在恒温槽中恒温。 图23-1测量线路图
开启电磁搅拌的电源使溶液在设定的温度下恒温至少10min。在以下系列实验过程中尽量使搅拌子的位置和转速保持一致。
(3)通过计算机使电化学分析仪进入Windows工作界面。
3.测量
(1)被测溶液在指定温度下恒温足够长时间(至少10min),点击工具栏里的运行键,实验即刻开始,屏幕上会显示电位一时间曲线(同时也分别显示电位和时间的数值),此时的曲线应该为一平线。60s(或基线平坦)后将预先已恒温的10 mL溴酸钾溶液倒入电解池中。此时曲线(电位)会发生突跃,同时注意溶液颜色的变化。经过一段时间的“诱导”,开始振荡反应,此后的曲线呈现有规律的周期变化(如图23-2所示),实验结束后给实验结果取个文件名存盘。
(2)将恒温槽温度调至32℃,取出电极洗净 图23-2振荡曲线图
电解池和所有用过的电极,然后重复上述步骤进行测量。分别每间隔2℃测定一条曲线,至少测量六个温度下的曲线。
(3)如有兴趣,在测量最后一条曲线前将参数改成Run Time(sec)=3000S或更长一些,从实验结果看化学振荡反应的“兴衰”。
五、思考题
1.影响诱导期、周期及振荡寿命的主要因素有哪些?
2.为什么在实验过程中应尽量使搅拌子的位置和转速保持一致?
六、注意事项及其它说明
1.所用的饱和甘汞电极与溶液之间必须用1 mol·L-1H2S04盐桥隔离。
2.每次实验完毕后必须将所有用具冲洗干净。
3.大多数反应在所研究的一定温度范围内是符合阿仑尼乌斯公式的,通常称复杂反应的活化能为表观活化能。
实验二十四 溶胶聚沉值的测定
一、实验目的
1.了解溶胶聚沉方法;
2.了解溶胶的保护;
3.掌握聚沉值的测定及其一些规律。
二、实验主要设备及使用要求
电炉;移液管;移液管;容量瓶
三、实验原理、方法和手段
胶体有巨大的表面和吸附作用,使胶粒往往都是带电的,如用FeCl3水解得到的Fe(OH)3,胶体其胶粒都是带正电的。
由于在同一条件下制得的胶粒都带有同一类型的电荷,胶粒之间存在着一种静电斥力,就使得它难于继续碰撞而长大,这是胶粒能较稳定存在之根源。
若改变溶胶生成的条件,如向溶胶中加入电解质,以减少或消除胶粒的电荷,胶粒较易于碰撞而变成大颗胶粒而沉降下来,这种过程称为溶胶的聚沉。
在一定的温度和压力条件下,要使溶胶在一定时间内发生明显的聚沉,所需电解质的最低浓度称聚沉值,单位为毫摩尔/升。
聚沉值的大小跟胶体性质和它所处条件有关。
温度升高,聚沉值减少,甚至有的胶体只用升高温度的方法来聚沉。
在温度一定时,聚沉值与电解质中被吸附的离子价(电解质中与胶粒电荷相反的离子)价数有关,其离子的价数越高,聚沉值越少,若价数相同时胶粒对有机离子的吸附比无机离子更为容易,故有机离子的聚沉值也就要小。
亲液胶体(明胶)加入到憎液胶体[Fe(OH)3溶液]中起保护作用。
四、实验内容与步骤
1.溶胶的制备
用量筒量取50ml蒸馏水,放入100ml的烧杯中,加热至沸腾,用移液管吸取2.5ml10%的FeCl3溶液,加到沸水中,得到棕红色的Fe(OH)3溶胶,冷却后备用。
2.溶胶聚沉值的初测定――近似聚沉值Cˊ
分两步进行,第一步测定近似聚沉值Cˊ,第二步测准确聚沉值C。
取试管6支,编号为1,2,3,4,5和对照,在1号试管注入10ml2.5MNaCl,在2-5号和对照等五支试管中各加入蒸馏水9ml,然后从1号试管中取1ml NaCl放入2号试管中,摇匀后,又从2号试管中取出1ml溶液放入3号试管中,依次从上一编号试管吸取1ml NaCl溶液,直至5号试管,最后从5号管中取出1ml溶液弃之。用移液管向上述1-5号试管及对照管中各加入已净化好的Fe(OH)3溶胶1ml,摇匀后,立即记下时间,静置15min,观察1-5号管的聚沉情况,与对照管进行比较,找出最后一支有沉淀的管子,该管的浓度即为近似聚沉值Cˊ,并记入表格内。
溶胶聚沉值的测定
编号
| 1
| 2
| 3
| 4
| 5
| 对照
|
2.5MNaCl(ml)
| 10.00
|
|
|
|
|
|
蒸馏水(ml)
|
| 9.00
| 9.00
| 9.00
| 9.00
| 9.00
|
摇匀后
| 1ml→
| 1ml→
| 1ml→
| 1ml→弃去
|
|
溶胶Fe(OH)3(ml)
| 1.00
| 1.00
| 1.00
| 1.00
| 1.00
| 1.00
|
|
|
|
|
|
|
|
|
|
|
|
3.溶胶聚沉值的精确测定
制备浓度Cˊ的NaCl溶液50ml。由原2.5MNaCl稀释而得,根据C1V1= CˊVˊ可求得V1的ml数。准确量取2.5MNaCl溶液V1ml至50ml的容量瓶中,用蒸馏水稀释至刻度,摇匀即得浓度为Cˊ的溶液。
在1)的1-5号试管中,保留具有近似聚沉值Cˊ浓度的试管,定它为Ⅰ试管,另取4支试管,编号为Ⅱ、Ⅲ、Ⅳ、Ⅴ,加上原对照一支又得一组6支管的排列。在Ⅱ-Ⅴ号试管中分别加入8、6、4、2ml的浓度为Cˊ的NaCl溶液,再向上述各管中加入1、3、5、7ml蒸馏水,摇匀后各加入1ml Fe(OH)3溶胶,摇匀后静置15min,找出最后一支有沉淀的管子,
编号
| Ⅰ
| Ⅱ
| Ⅲ
| Ⅳ
| Ⅴ
| 对照
|
CˊNaCl(ml)
|
| 8.00
| 6.00
| 4.00
| 2.00
|
|
蒸馏水(ml)
|
| 1.00
| 3.00
| 5.00
| 7.00
| 9.00
|
溶胶Fe(OH)3(ml)
|
| 1.00
| 1.00
| 1.00
| 1.00
| 1.00
|
用这支管子中的NaCl的浓度Cn和相邻未沉淀的管子NaCl浓度Cn+1求出平均值,即得所求的准确聚沉值C,即C=1/2(Cn+ Cn+1)。把所观察的现象和计算结果列入表中。
4.用上述完全相同的手续,测定Na2SO4对Fe(OH)3溶胶的聚沉值。
五、思考题
1.什么是聚沉值?
2.做好本实验的关键是什么?
实验二十五 溶液吸附法测定比表面积
一、实验目的
1.用溶液吸附法测定颗粒活性炭的比表面。
2.了解溶液吸附法测定比表面的基本原理。
3.了解721型分光光度计的基本原理并熟悉使用方法。
二、实验主要设备及使用要求
721型分光光度计
三、实验原理、方法和手段
(1)比表面是指单位质量(或单位体积)的物质所具有的表面积,其数值与分散粒子大小有关。测定固体物质比表面的方法很多,常用的有BET低温吸附法、电子显微镜法和气相色谱法等,不过这些方法都需要复杂的装置,或较长的时间。而溶液吸附法测定固体物质比表面,仪器简单,操作方便,还可以同时测定许多个样品,因此常被采用,但溶液吸附法测定结果有一定误差。其主要原因在于:吸附时非球型吸附层在各种吸附剂的表面取向并不一致,每个吸附分子的投影面积可以相差很远,所以,溶液吸附法测得的数值应以其它方法校正之。然而,溶液吸附法常用来测定大量同类样品的相对值。溶液吸附法测定结果误差一般为10%左右。
(2)水溶性染料的吸附已广泛应用于固体物质比表面的测定。在所有染料中,次
甲基蓝具有最大的吸附倾向。研究表明,在大多数固体上,次甲基蓝吸附都是单分子层,即符合朗格缪尔型吸附。但当原始溶液浓度较高时,会出现多分子层吸附,而如果吸附平衡后溶液的浓度过低,则吸附又不能达到饱和,因此,原始溶液的浓度以及吸附平衡后的溶液浓度都应选在适当的范围内。本实验原始溶液浓度为0.2%左右,平衡溶液浓度不小于0.1%。
(3)根据朗格缪尔单分子层吸附理论,当次甲基蓝与活性炭达到吸附饱和后,吸附与脱附处于动态平衡,这时次甲基蓝分子铺满整个活性粒子表面而不留下空位。此时吸附剂活性炭的比表面可按式(1)计算:
(1)
式中,S0为比表面(m2·kg-1);C0为原始溶液的质量分数;C为平衡溶液的质量分数;G为溶液的加入量(kg); W为吸附剂试样质量(kg);2.45×106是1kg次甲基蓝可覆盖活性炭样品的面积(m2·kg-1)。
(4)次甲基蓝分子的平面结构如图25-1所示。阳离子大小为1.70×10-10m×76×10-10m×325×10-10m。次甲基蓝的吸附有三种趋向:平面吸附,投影面积为1.35×10-18m2;侧面吸附,投影面积为7.5×10-19m2;端基吸附,投影面积为39.5×10-19m2。对于非石墨型的活性炭,次甲基蓝可能不是平面吸附,也不是侧面吸附,而是端基吸附根据实验结果推算,在单层吸附的情况下,1mg次甲基蓝覆盖的面积可按2.45m2计算。
(5)本实验溶液浓度的测量是借助于分光光度计来完成的。根据光吸收定律,当入射光为一定波长的单色光时,某溶液的光密度与溶液中有色物质的浓度及溶液的厚度成正比,
即
A=LogI0/I=KCL
图25-2 溶液吸收曲线
其中: A——吸光度;
I——透射光强度;
I0 ——入射光强度;
K——吸收系数;
C——溶液浓度;
L——溶液的光径长度。
一般说来,光的吸收定律能适用于任何波长的单色光,但对于一个指定的溶液,在不同的波长下测得的吸光度不同。如果把波长λ对吸光度A作图,可得到溶液的吸收曲线,如图25-2所示。
为了提高测量的灵敏度,工作波长应选择在吸光度A值最大时所对应的波长。对于次甲基蓝,本实验所用的工作波长为665nm。
实验首先测定一系列已知浓度的次甲基蓝溶液的吸光度,绘出A—C工作曲线,然后测定次甲基蓝原始溶液及平衡溶液的吸光度,再在A—C曲线上查得对应的浓度值,代入(1)式计算比表面。
四、实验内容与步骤
1.活化样品:将颗粒活性炭置于瓷坩锅中,放入马弗炉内,500℃下活化1h(或在真空烘箱中300℃下活化1h),然后放入干燥器中备用。
2.取两只带塞磨口锥形瓶,分别加入准确称量过的约0.2g的活性炭(两份尽量平行),再分别加入50g(50mL)0.2%的次甲基蓝溶液,盖上磨口塞,轻轻摇动,其中一份放置1h,即为配制好的平衡溶液,另一份放置一夜,认为吸附达到平衡,比较两个测定结果。
3.配制次甲基蓝标准溶液:用移液管分别量取5mL、8mL、11mL 0.01%标准次甲基蓝溶液置于1000mL容量瓶中,用蒸馏水稀释至1000mL,即得到5×10-6、8×10-6、11×10-6三种浓度的标准溶液。
4.平衡溶液处理:取吸附后平衡溶液约5mL,放入1000mL容量瓶中,用蒸馏水稀释至刻度。5.选择工作波长:对于次甲基蓝溶液,吸附波长应选择655nm,由于各台分光光度计波长
略有差别,所以,实验者应自行选取工作波长。用5×10-6标准溶液在600nm~700nm范围测量吸光度,以吸光度最大时的波长作为工作波长。
6.测量溶液吸光度:以蒸馏水为空白溶液,分别测量5×10-6、8×10-6、11×10-6三种浓度的标准溶液以及稀释前的原始溶液和稀释后的平衡溶液的吸光度。每个样品须测得三个有效数据,然后取平均值。
五、思考题
1.为什么次甲基蓝原始溶液浓度要选在0.2%左右,吸附后的次甲基蓝溶液浓度要在0.1%左右?若吸附后溶液浓度太低,在实验操作方面应如何改动?
2.用分光光度计测定次甲基蓝溶液浓度时,为什么要将溶液稀释到1/1000000浓度才进行测量?
3.如何才能加快吸附平衡的速度?
4.吸附作用与哪些因素有关?
六、注意事项及其它说明
1.测定溶液吸光度时,须用滤纸轻轻擦干比色皿外部,以保持比色皿暗箱内干燥。
2.测定原始溶液和平衡溶液的吸光度时,应把稀释后的溶液摇匀再测。
3.活性炭颗粒要均匀,且三份称重应尽量接近。
实验二十六电渗
一、实验目的
1.通过电渗法测定SiO2对水的电势,掌握电渗法测定电势的基本原理和技术。
2.加深理解电渗是胶体中液相和固相在外电场作用下相对移动而产生的电性现象。
二、实验主要设备及使用要求
电渗仪;直流电源(200~1000V);电导率仪


图26-1 电渗仪结构示意图
三、实验原理、方法和手段
电渗属于胶体的电动现象。电动现象是指溶胶粒子的运动与电性能之间的关系。一般包括电泳、电渗、流动电位与沉降电位。电动现象的实质是由于双电层结构的存在,其紧密层和扩散层中各具有相反的剩余电荷,在外电场或外加压力下,它们发生相对运动。
电渗是指在电场作用下,分散介质通过多孔膜或极细的毛细管而定向移动的现象。
若知道液体介质的粘度,介电常数,电导率,只要测定在电场作用下通过液体介质的电流强度I,和单位时间内液体流过毛细管的流量v,可根据下式求出电势。
四、实验内容与步骤
1.洗净电渗仪,将80-100目的二氧化硅与蒸馏水拌成的糊状物注入A管。
2.从电极管口注入蒸馏水,直至浸没铂丝电极为止,插好铂丝电极。
3.用胶头滴管在负极端挤一个气泡至刻度毛细管的一端。
4.在电渗仪的两铂丝电极间接上直流电源,调节电源电压,使电渗时毛细管中气泡从一端刻度至另一段刻度行程时间约为20s。然后准确测定此时间。利用换向开关,改变电渗方向,分别测量正反向电渗时的流量和电流强度值五次。
改变电源电压,把毛细管中气泡的行程时间分别改为15s,25s,按上述方法分别测量相应的流量和电流强度值。
5.最后拆去电渗仪电源,用电导率仪测定电渗仪中蒸馏水的电导率。
五、思考题
1.为什么说刻度毛细管中气泡在单位时间内移动的体积就是单位时间内流过试样室A管的液体量?
2.电渗测量时,连续通电使溶液发热,会造成什么后果?
3.固体粉末样品粒度太大,电渗测定的结果重现性差,其原因何在?
六、注意事项及其它说明
1.如用蒸馏水实验不成功,建议用0.001mol/LNaCl溶液代替。
2.电源电压不能太高,否则溶液本身电解越强,影响测量结果。
3.计算SiO2对水的电势时,注意各物理量的单位。在法定计量单位实行之后,计算公式中不应有。
实验二十六Fe(OH)3胶体电动电位的测定—界面电泳法
一、实验目的
1.理解电动电势ζ的物理意义,掌握用电泳法测定ζ电势的原理和技术。
2.加深理解在外电场作用下胶粒与周围介质作相对运动时产生的动电现象。
二、实验主要设备及使用要求
电泳仪;电泳管

图26-1电泳测定装置
三、实验原理、方法和手段
本试验采用Fe(OH)3胶体进行电泳试验,Fe(OH)3溶胶用水解凝聚法制备,制备过程中所涉及的化学反应过程如下:
(1)在沸水中加入FeCl3溶液: FeCl3+ 3H2O = Fe(OH)3+ 3HCl
(2)溶胶表面的Fe(OH)3会再与HCl反应: Fe(OH)3+ HCl = FeOCl + 2 H2O
(3)FeOCl离解成FeO+和Cl-离子。胶团结构为: [Fe(OH)3]m·nFeO+·(n-x)Cl-]x+·xCl-
胶体溶液(溶胶)是由分散相线度在10-9~10-7m的高分散
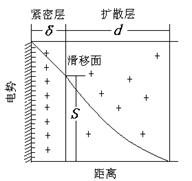 多相体系。胶核大多是分子或原子的聚集体,因选择性地吸附介质中的某种离子(或自身电离)而带电,介质中存在的与吸附离子电荷相反的离子称为反离子,反离子中有一部分因静电引力(或范德华力)的作用,与吸附离子一起紧密地吸附于胶核表面,形成紧密层。于是胶核、吸附离子和部分反离子(即紧密层)构成了胶粒。反离子的另一部分由于热扩散分布于介质中,故称为扩散层,见图26-2。 图26-2双电层示意图
多相体系。胶核大多是分子或原子的聚集体,因选择性地吸附介质中的某种离子(或自身电离)而带电,介质中存在的与吸附离子电荷相反的离子称为反离子,反离子中有一部分因静电引力(或范德华力)的作用,与吸附离子一起紧密地吸附于胶核表面,形成紧密层。于是胶核、吸附离子和部分反离子(即紧密层)构成了胶粒。反离子的另一部分由于热扩散分布于介质中,故称为扩散层,见图26-2。 图26-2双电层示意图
紧密层与扩散层交界处称为滑移面(或Stern面),显然紧密层与介质内部之间存在电势差,该电势差称为ζ电势。在电场中胶粒会向异号电极移动,即电泳现象,在特定的电场中,ζ电势的大小取决于胶粒的运动速度,故ζ电势又称为电动电势。
溶胶之所以在一定条件下能相对稳定的存在,主要原因之一就是体系中胶粒带有相同的电荷,彼此之间排斥不致聚集。胶粒带的电荷越多,ζ电势越大,胶体体系越稳定。因此,ζ电势大小是衡量溶胶稳定性的重要参数。
ζ电势的测定方法有多种,利用电泳现象可测定ζ电势。电泳法又分为宏观法和微观法,前者是将溶胶置于电场中,观察溶胶与另一不含溶胶的导电液(辅助液)间所形成的界面的移动速率;后者是直接观测单个胶粒在电场中的泳动速率。对高分散或过浓的溶胶只能用宏观法;对颜色太浅或浓度过稀的溶胶只能用微观法。
本实验采用宏观法测定Fe(OH)3溶胶的ζ电势,其ξ电势可按下式计算:
式中η、ε为测量温度下介质的粘度(Pa·s)和介电常数;u为胶粒电泳的相对移动速率(m·s-1);(V /l)是电位梯度(V·m-1),V是两电极间电位差(V);l为两电极间距离(m);K是与胶粒形状有关的常数,球形粒子为6,棒状粒子为4,对Fe(OH)3K值为4。
四、实验内容与步骤
1.胶体的制备:
用水解法制备Fe(OH)3溶胶:在250mL烧杯中,加入100mL蒸馏水,加热至沸,慢慢滴入5-10mL (20%) FeCl3溶液,并不断搅拌,加毕继续保持沸腾5min,即可得到红棕色的Fe(OH)3溶胶,其结构式可表示为:
{m[Fe(OH)3]nFeO+(n-x)Cl-}x+xCl-。
在胶体体系中存在过量的H+、Cl-等离子需要除去。
2. 胶体的纯化:
(1)制备半透膜在一个内壁洁净、干燥的250mL锥形瓶中,加入约10mL火棉胶液,小心转动锥形瓶,使火棉胶液粘附在锥形瓶内壁上形成均匀薄层,倾出多余的火棉胶于回收瓶中。此时锥形瓶仍需倒置,并不断旋转,待剩余的火棉胶流尽,使瓶中的乙醚蒸发至已闻不出气味为止(此时用手轻触火棉胶膜,已不粘手)。然后再往瓶中注满水,(若乙醚未蒸发完全,加水过早,则半透膜发白)浸泡10min。倒出瓶中的水,小心用手分开膜与瓶壁之间隙。慢慢注水于夹层中,使膜脱离瓶壁,轻轻取出,在膜袋中注入水,观察有否漏洞。制好的半透膜不用时,要浸放在蒸馏水中。
(2)纯化Fe(OH)3溶胶 用热渗析法纯化Fe(OH)3溶胶:将制得的Fe(OH)3溶胶,注入半透膜内用线拴住袋口,置于800mL的清洁烧杯中,杯中加蒸馏水约300mL,维持温度在60℃左右,进行渗析。半小时后,取溶胶用1% AgNO3及1%KCNS溶液检查是否存在Cl-及Fe3+,如果仍存在,应继续换水继续渗析,直到检查不出为止,将纯化过的Fe(OH)3溶胶移入一清洁干燥的100mL小烧杯中待用。
3. 清洁电泳管加入适量的Fe(OH)3溶胶,在U型管两液面分别缓慢加入KCl辅助液,使形成清晰界面。
4. 将石墨电极插入KCl辅助液层中,两电极以并联的形式与电泳仪连接。插入后使两电极垂直,而且在KCl辅助液层中深度相等,记下胶体界面的初试高度位置。
5. 连接电源,同时开动计时器记时,连续电泳30min左右以后记下胶体液面上升的距离和电压的读数。
6. 切断电源,用软线测量电泳管中两电极在胶体中的距离,测量4~5次,取平均值。
7. 实验结束,拆除装置,清洗电泳管。
五、思考题
1.如果电泳仪事先没有洗干净,管壁上残留有微量的电解质,对电泳的测量结果有何影响?
2.电泳速率的快慢与哪些因素有关?
六、注意事项及其它说明
1.半透膜制备:一定要使整个锥形瓶的内壁上均匀地附着一层火棉胶液,在取出半透膜时,一定要借助水的浮力将膜托出。
2.Fe(OH)3溶胶制备:FeCl3一定要逐滴加入,并不断搅拌。
3.Fe(OH)3溶胶纯化时,换水后要渗析一段时间再检查Fe3+及Cl-的存在。
4.电泳过程中要保持界面清晰(减少电泳管晃动)。
实验二十八 粘度法测定水溶性高聚物的分子量
一、实验目的
1.了解粘度的物理意义、测定原理和方法。
2.了解粘度法测定高聚物摩尔质量的基本原理和公式。
3.掌握用乌氏(Ubbelohde)粘度计测定高聚物溶液粘度的原理与方法。
4.测定聚乙二醇的摩尔质量。
二、实验主要设备及使用要求
恒温槽;乌氏(乌贝路德)粘度计
三、实验原理、方法和手段
当流体受外力作用产生流动时,在流动着的液体层之间存在着切向的内部摩擦力。如果要使液体通过管子,必须消耗一部分功来克服这种流动的阻力。在流速低时管子中的液体沿着与管壁平行的直线方向前进,最靠近管壁的液体实际上是静止的,与管壁距离愈远,流动的速度也愈大。
流层之间的切向力f与两层间的接触面积A和速度差Δv成正比,而与两层间的距离Δx成反比:
式中,η称为液体的粘度系数,简称粘度。在国际单位制(SI)中量刚为:kg·m-1·s-1或Pa·s表示。
高聚物相对分子质量不仅反映了高聚物分子的大小,而且直接关系到它的物理性能,是个重要的基本参数。与一般的无机物或低分子的有机物不同,高聚物大多是摩尔质量大小不同的大分子混合物,所以通常所测高聚物摩尔质量是一个统计平均值。
测定高聚物摩尔质量的方法很多,而不同方法所得平均摩尔质量也有所不同。比较起来,粘度法(毛细管法)设备简单,操作方便,并有很好的实验精度,是常用的方法之一。用该法
表27-1常用粘度术语的物理意义
符号
| 名称与物理意义
|
η0
| 纯溶剂的粘度,溶剂分子与溶剂分子间的内摩擦表现出来的粘度。
|
η
| 溶液的粘度,溶剂分子与溶剂分子之间、高分子与高分子之间和高分子与溶剂分子之间三者内摩擦的综合表现。
|
ηr
| 相对粘度,ηr=η/η0,溶液粘度对溶剂粘度的相对值。
|
ηsp
| 增比粘度,ηsp= (η-η0) /η0=η/η0–1 =ηr– 1,反映了高分子与高分子之间,纯溶剂与高分子之间的内摩擦效应。
|
ηsp/C
| 比浓粘度,单位浓度下所显示出的粘度。
|
[η]
| 特性粘度,,反映了高分子与溶剂分子之间的内摩擦。其单位是浓度c单位的倒数。
|
求得的摩尔质量称为粘均摩尔质量。
粘度法测高聚物溶液摩尔质量时,常用名词的物理意义,如表27-1所示。在足够稀的高聚物溶液里,ηSP/c与c和lnηr/c与c之间分别符合下述经验公式:
 ,
,
式中,κ和β分别称为Huggins和Kramer常数。这是两个直线方程,因此我们获得[η]的方法如左图所示。一种方法是以ηSP/c对c作图,外推到c→0的截距值;另一种是以lnηr/c对c作图,也外推到c→0的截距值,两条线应会合于一点, 图27-1外推法求特性粘度[η]
这也可校核实验的可靠性(见图27-1)。
在一定温度和溶剂条件下,特性粘度[η]和高聚物摩尔质量M之间的关系通常用带有两
 参数的个Mark-Houwink经验方程式来表示:
参数的个Mark-Houwink经验方程式来表示:
式中,M为粘均摩尔质量;K为比例常数;α是与分子形状有关的经验参数。K和α值与温度、聚合物、溶剂性质有关,也和分子量大小有关。K值受温度的影响较明显,而α值主要取决于高分子线团在某温度下,某溶剂中舒展的程度,其数值介于0.5~1之间。K与α的数值可通过其它绝对方法确定,例如渗透压法、光散射法等,由粘度法只能测定[η]。
对于本实验,已知:35℃时,聚乙二醇,,。
可以看出高聚物摩尔质量的测定最后归结为特性粘度[η]的测定。本实验采用毛细管法测定粘度,通过测定一定体积的液体流经一定长度和半径的毛细管所需时间而获得。所使用的乌氏粘度计如图27-2所示,当液体在重力作用下流经毛细管时,其遵守泊肃叶(Poiseuille)定律:
式中,η为液体的粘度;ρ为液体的密度;L为毛细管的长度;r为毛细管的半径;t为V体积液体的流出时间;h为流过毛细管液体的平均液柱高度;V为流经毛细管的液体体积;m为毛细管末端校正的参数(一般在时,可以取m=1)。
对于某一只指定的粘度计而言,许多参数是一定的,因此上式可以改写成:
式中,B<1,当流出的时间t在2min左右(大于100s),该项(亦称动能校正项)可以忽略,即η=Aρt。
又因通常测定是在稀溶液中进行(C<1×10-2g·cm-3),溶液的密度和溶剂的密度近似相等,因此可将ηr写成:
式中,t为测定溶液粘度时液面从a刻度流至b刻度的时间;t0为纯溶剂流过的时间。
所以通过测定溶剂和溶液在毛细管中的流出时间,可以测定相对粘度以及增比粘度),,通过、对c作图外推可得。根据公式: ,有了,则分子量可求出。
四、实验内容与步骤
1.将粘度计(见图2-30-1)用洗液、自来水和蒸馏水洗干净,每次都要注意反复流洗毛细管
部分,用乙醇润洗,然后烘干备用。
2.调节恒温槽温度至(35.0± 0.1)℃,将乌氏粘度计在恒温槽中恒温10分钟(注意要垂直放置)。纯水及标样(聚乙二醇溶液事先已配制好了,浓度为1g/100ml)于35℃水中恒温。
3.溶剂流出时间t0的测定:向粘度计中加入纯水20ml,恒温2分钟,进行测定。测定方法如下:使C管不通气,在B管处用洗耳球将溶剂从F球经D球、毛细管、E球抽至G球2/3处,让C管通大气,此时D球内的溶液即回入F球,使毛细管以上的液体悬空。毛细管以上的液体下落,当液面流经a刻度时,立即按停表开始记时间,当液面降至b刻度时,再按停表,测得刻度a、b之间的液体流经毛细管所需时间。重复这一操作至少三次,相差不大于0.3s,取三次的平均值为t0。
4.溶液流出时间的测定:烘干乌氏粘度计,由A管注入注入10ml标样,于35℃恒温2分钟,向粘度计中注入2ml水,使B管不通气,在C管处用洗耳球打气,使溶液混合均匀,浓度记为C1,恒温2min,测定流出时间三次,并取平均值为t1。
5.接着再向粘度计中注入3ml水,用洗耳球将液体混匀(恒温2分钟),使溶液浓度分别为C2,并多次抽洗粘度计的E球和G球以及粘度计的毛细管部分后,测定流出时间三次,并取平均值为t2。
6.重复5的操作,只是加水量分别为5、10ml,将溶液稀释,使溶液浓度分别为C3、C4,用同法测定每份溶液流经毛细管的时间t3、t4。应注意每次加入水后,要充分混合均匀,并多次抽洗粘度计的E球和G球,使粘度计内溶液各处的浓度相等。
7.实验完毕后,关闭电源,小心地清洗粘度计三次,然后用纯水注满粘度计(要求水面覆盖到粘度计的毛细管之上)。关闭电源,原始数据记录本交给老师签字。
五、思考题
1.乌氏粘度计中支管C有何作用?除去支管C是否可测定粘度?
2.乌氏粘度计的毛细管太粗或太细有什么缺点?
3.为什么用[η]来求算高聚物的摩尔质量?它和纯溶剂粘度有无区别?
4.分析ηSP/c-c及lnηr/c-c作图缺乏线性的原因?
六、注意事项及其它说明
1.在配制高聚物溶液时必须保证其完全溶解,否则会影响溶液起始浓度,而导致结果偏低。
2.粘度计必须洁净,高聚物溶液中若有絮状物不能将它移入粘度计中,过滤后方可使用。
3.本实验中溶液的稀释是直接在粘度计中进行的,因此每加入一次溶剂进行稀释时必须混合均匀,并抽洗E球和G球。
4.实验过程中恒温槽的温度要恒定,溶液每次稀释恒温后才能测量。
5.粘度计要垂直放置,实验过程中不要振动粘度计,否则影响结果的准确性。
6.毛细管粘度计的毛细管内径选择,可根据所测物质的粘度而定,内径太细,容易堵塞,内径太粗,测量误差太大,一般选择测水时流经毛细管的时间大于100s,在120s左右为宜。
7.若作图时两直线不能在纵轴上交于一点,则取直线的截距作为。
实验二十九 配合物磁化率的测定
一、实验目的
1.掌握用Gouy法测定配合物磁化率的原理和方法
2.通过配合物磁化率的测定,计算其中心金属离子的未成对电子数,并判断配合物中配键
的键型
二、实验主要设备及使用要求
古埃磁天平,电子太平
三、实验原理、方法和手段
1.磁(介)质的摩尔磁化率χM
磁(介)质分为:铁磁质(Fe、Co、Ni及其化合物)和非铁磁质。
非铁磁质分为:反磁质(即反磁性物质)和顺磁质(即顺磁性物质),顺磁质中含有未成对电子。
在不均匀磁场中,反磁质受到的磁场作用力很小,该作用力由磁场强度大的地方指向磁场强度小的地方。所以,本实验中反磁质处于不均匀磁场中时的质量比无外磁场时的稍小一点;而顺磁质受到的磁场作用力较大,作用力由磁场强度小的地方指向磁场强度大的地方。即,本实验中顺磁质处于不均匀磁场中时的质量比无外磁场时的质量有明显增大。
化学上人们感兴趣的是非铁磁质。非铁磁质中的反磁质具有反磁化率,顺磁质同时具顺磁化率和反磁化率,但其顺磁化率(正值)远大于其反磁化率(负值)。所以,对顺磁质而言,其摩尔磁化率:
χM= χμ(摩尔顺磁化率)+ χ0(摩尔逆磁化率)≈ χμ
而 χμ= (在本实验中χμ的单位为:cm3·mol-1)
上式中,g为重力加速度(SI单位为:m·s-2),H为磁场强度(单位为:Oe,读作“奥斯特”),在本实验的计算中其值也可消去,亦不必考虑其取值的大小及单位;M为样品的摩尔质量,在本实验的计算中其单位取g/mol;h为样品管中所装样品粉末的高度,在本实验的计算中其单位取cm;WH为有外加磁场时“样品+试管”的质量与“空试管”的质量之差,单位为g;WO为无外加磁场时“样品+试管”的质量与“空试管”的质量之差,单位为g。
2.磁场强度H的标定
若已知某样品的磁化率,则可通过实验利用下式求出对应的磁场强度。
χM= (cm3·mol-1)
同理,若已知某样品的比磁化率(即单位质量磁介质的磁化率)χm(m3·kg–1,或cm3·g-1),则亦可通过实验利用下式求出对应的磁场强度。
χm=
已知莫尔盐(六水合硫酸亚铁铵晶体)的比磁化率仅是热力学温度T/K的函数,具体关系为:
χm=(m3·kg–1)(其中,热力学温度T的单位K已预先消去了)
而根据SI制与CGS制的换算,有:,所以,
χm= (cm3·g–1)
因此,只要在一定温度下,用莫尔盐进行测定,即可由下式求出磁场强度的平方。
表面上看,本实验似乎需要求出磁场强度的平方用于后续计算,但实际上,在具体的数据处理中我们并不由上式求出具体数值,因为有些量可以消去(见后面的讨论)。式中W的意义见实验步骤部分。
3.样品的摩尔磁化率χM的测定
χM≈ χμ= (式中M为样品的摩尔质量,单位g/mol)
=
=(注意:顺磁质χM>0,反磁质χM<0 )
可见,只要测出样品管中有关样品(两个样品以及莫尔盐)的高度,样品在有外磁场时的质量、样品在无外磁场时的质量,即可求出样品的摩尔磁化率,而不需计算磁场强度的平方。
4.求样品的永久磁矩μ
将上述计算所得的χM(cm3·mol–1)代入下式,可得永久磁矩的平方值。
χM=χM()
5.样品的未成对电子数n的计算
式中,是电子的磁矩(玻尔磁子)= 9.27×10-21erg/Oe。
四、实验内容与步骤
1.阅读古埃磁天平的使用说明,理解其使用方法,复习电子天平的操作方法。
2.先将磁天平上的电流旋钮旋至电流为零的位置,在打开磁天平上的电源总开关。通过电
流强度的增减调节磁天平的电磁铁的磁场强度时,应缓慢地增大或减小电流。增大磁场
时,要由小到大缓慢地增大电流;关闭磁场时,要由大到小缓慢地减小电流。
3.调节电流大小使古埃磁天平电磁铁的磁场强度为300mT左右,再将空试管吊放于磁场
的适当位置(参见图1),测定有磁场时空试管的质量mH,管,再将电流调为零,称量无
磁场时空试管的质量m0,管。因使用电子天平称重,所以每次测定的读数只需读数一次。
4.装入样品莫尔盐粉末,并用玻棒将试管上部的固体轻轻压平,以方便量出样品的高度。然后调节电流大小使磁天平电磁铁的磁场强度达到300mT左右,小心地将已装样的试管挂到电子天平下的吊环上,称量有磁场时“莫尔盐+试管”的质量mH,管+莫;再将电流调为零,称量无磁场时“莫尔盐+试管”的质量mH,管+莫。一个样品用完后应按要求倒回到原试剂瓶中(切不可倒错试剂瓶),或丢弃于指定的其它容器中,然后先用自来水洗净试管,再用蒸馏水淌洗两次,倒去试管中的余水后,向试管中加入约1毫升无水乙醇将试管淌洗一遍,淌洗后将乙醇倒如指定的试剂瓶中,而后用电吹风吹干试管用于另一个样品的称量。
5.重复第4步的操作,分别测定硫酸亚铁晶体、亚铁氰化钾晶体(均为粉末)在有磁场时“样品+试管”的质量mH,管+样、无磁场时“样品+试管”的质量mH,管+样。
6.可用于数据处理的W值:
WH,莫= mH,管+莫 - mH,管 W0,莫= m0,管+莫 - m0,管
WH,样= mH,管+样 - mH,管 W0,样= m0,管+样 - m0,管
7.实验完毕后,将电流旋钮缓慢调到零,再关闭电源开关,打扫小组实验台面的卫生,抄录一份原始实验数据记录,交教师签字后离开实验室。
五、思考题
1.实验过程中,为什么每个实验小组始终只能使用同一支试管?
2.摩尔盐的什么特点使得它可用于标定实验中的磁场强度?
六、注意事项及其它说明
1.装样时要一边装样一边在桌面上轻轻地震动试管,使样品紧密均匀地填实试管,当样品表面距管口约0.5cm时停止装样,最后用各样品专用的小玻棒将管内表面的样品轻轻压平。
2.由于试管是被挂载于天平托盘底部所连接的一根细绳下面的挂钩上的,而电子天平的最大称重量只有约100克,所以,要特别注意保护电子天平,任何时候都严禁向下拉动细绳。挂载试管时要一手将细绳向上提起,另一手将试管挂于挂环上,然后再将细绳连同试管一起缓慢、轻轻地放下。取下试管时,则要首先将细绳向上提起,另一手取走试管,然后缓慢地放下细绳。操作中还要尽量减少试管的摆动,以保护天平。而且,挂载试管的细绳不应与任何物体接触。
3.称量过程中,试管应与地面垂直,试管底部则要刚好位于两快电磁铁的上下位置和左右
位置的正中央。